
 Enzymes and Surface Water
Enzymes and Surface Water
Wiggins P1
1*Retired from the Department of Medicine,University of Auckland, New Zealand, and from Genesis Research and Development Company, Auckland, New Zealand.
Correspondence: p.wiggins@paradise.net.nz
Key Words: allostery, protein folding, water
Received 12 February 2009; revised 17 March 2009; accepted 9 April 2009. Published 1 July 2009. Available online 1 July 2009.
Summary
The broad OH-stretch band of the infra red spectrum of liquid water is shown to comprise two overlapping bands peaking at 3250 cm-1 (the value in ice, and, presumably, strongly bonded water) and 3635 cm-1 (presumably weakly bonded water). The spectra also reveal the coexistence of zones of LDW and HDW in small-pored polymeric matrices. Possible mechanisms of reactions catalyzed by these zones of water associated with enzymes are described. There is a crucial functional connection between the force that drives folding of an enzyme and reactions that it catalyses. When water can move to abolish osmotic pressure gradients created by selective uptake of solutes into HDW or LDW, it does so with some decrease in the partition coefficients of the reactants. When water is prevented from moving, partition coefficients are unchanged, increased or transiently inverted. Examples of allostery and Michaelis-Menten kinetics (Matthews and van Holde, 1990) are given.
Article Outline
- Introduction
- Generation and Properties of the Zones of High Density Water (HDW) and Low Density Water (LDW)
- A Representative Reaction in HDW
- Apparent Saturation Kinetics
- Promoters and Inhibitors (Allostery)
- The Same Reaction Catalysed by an Enzyme that Partitions into LDW
- A Representative Reaction in LDW
- Cellulose Acetate Membranes
- Reaction in LDW with Swelling
- Reaction at a Charged Site
- Dangers of Charged Sites
- Oxygenation of Haemoglobin
- Haemoglobin
- CO2 and Biphosphoglycerate
- The Bohr Effect
- The Role of Aminoacid Sequences
- Conclusion
- References
- Discussion with Reviewers
Introduction
Amino acids are capable of only weak interactions such as dipole-dipole attractions, H-bonds and some attraction between positive and negative sites. It is hard to see, even with induced fit, what generates the extreme specificity of the assumed binding sites. When they were first proposed, there was no plausible alternative mechanism. This vacuum was not helped in the early 1970s by the polywater debacle, which erased water from the biochemical lexicon (Franks, 1981). Now, however, two kinds of surface water with different solvent properties do supply plausible alternatives of great specificity. They have been shown to exist in solutions and at surfaces in non-biological systems such as porous beads and desalination membranes. Other attributes of these different surface waters are discussed here to see to what extent they are consistent with the extensive experimental data on enzyme reactions.
Generation and Properties of the Zones of High Density Water (HDW) and Low Density Water (LDW)

Figure 1: a small solute (red) has dissolved in (a) “classical water,” (b) HDW, and (c) LDW. In each case,water immediately adjacent to the solute has a lower concentration (activity) than the same volume away from the solute. This activity gradient results in a pressure gradient, which is positive immediately adjacent to the solute and negative further away. a, nothing happens because “classical” water is assumed to be impervious to the low pressures encountered in osmotic systems; b, water immediately adjacent to the solute is already HDW and is not further affected; c, water immediately adjacent to the surface is converted to HDW, while water in the zone of negative pressure is already LDW and is not further affected.
Figure 1 shows that at a surface, such as that of a protein, there is a gradient of water activity (Wiggins, 2008a), low near the surface and higher further out. Water in the zone of higher activity tends to move in to increase its activity in the zone of low activity. Since it cannot do this, the water activity gradient becomes a pressure gradient, with positive pressure on water at the surface and negative pressure further out. Hitherto, this gradient has been interpreted as just two zones of water: high density water (HDW) immediately close to the surface and low density water (LDW) further out. When it comes to enzyme function, this sharp division lacks subtlety. The gradient of pressure is continuous: the composition of the water changes from highly enriched in HDW at the surface to highly enriched in LDW where the pressure is most negative. The zones of HDW and LDW at the surface, therefore, are not pure and can be manipulated. This modification is necessary in order to explain many experimental results with enzymes and with non-living systems such as small-pored polyamide beads, and solutions of dextran sulphate. These results will be shown as they become relevant.
A Representative Reaction in HDW
Most if not all active sites are in narrow clefts between domains of the folded protein. Presumably, water in those clefts consists of zones of enriched HDW and zones of enriched LDW, similar to those identified in small-pored polyamide beads (Wiggins, 1988) and cellulose acetate films (Wiggins and van Ryan, 1986), both of which have pores (1-2 nm in diameter). It will be assumed that the particular enzyme under consideration is one such, and that it catalyses the reaction
Let the enzyme be of such an amino acid composition that it partitions into HDW and induces extra LDW; and let the reaction be one that takes place in highly enriched HDW at the surface. Since this water is under positive pressure, solutes do not induce LDW as they do in Figure 1.Therefore HDW can ionize freely without producing the LDW which inhibits ionization in LDW/HDW (Wiggins, 2007). Its low viscosity and relatively high concentrations of H+ and OH- ions make it a powerful catalyst. The steps of the reaction are:
1. Uptake of A and B into HDW from an external concentration of CA and CB
2. Reaction, A + B = C + D
3. Release of products.
In order to reach the zone of HDW, A and B must pass through the zone of LDW for which they have low affinity. Since all four participants in the reaction are biomolecules, they consist of both moieties that partition into HDW and moieties that partition into LDW; so although A and B have lower affinity for LDW than for HDW, they are not totally excluded: they can diffuse across.
A general principle of osmotic theory says that if water can move to abolish an osmotic pressure gradient, it must and will. If water moves to abolish the osmotic pressure gradient created by uptake of A and B, it must move into the zone under positive pressure where A and B are in solution. In so doing, some LDW/HDW is converted to HDW. Folding of this protein was driven by the need to reduce the excess LDW induced by its elongated conformation. Production of excess HDW by uptake of A and B, therefore, cancels some of this excess LDW and the protein can relax and open its cleft a little: so, water can move in and therefore does. The partition coefficients are modest and the flux of water not great.
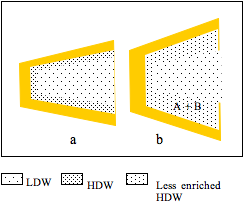
Figure 2: a, a cleft between two protein domains contains HDW at the surfaces and LDW in the centre; b, A and B partition into HDW; LDW/HDW moves into abolish the osmotic pressure gradient; most of this water converts to HDW because it is in the zone under positive pressure from the surface, but the extra water encroaches on the zone of negative pressure; its average enrichment with HDW decreases. The reaction (A + B = C + D) takes place and C and D diffuse out.
Figure 2 shows that uptake of A and B is accompanied by flux of water from the external solution of LDW/HDW into the zone of HDW. Since, however, the pressure gradient imposed by the surface has not changed, some extra water encroaches on the zone of negative pressure. This decreases the effective enrichment of HDW in the solution round A and B and, therefore, the partition coefficients of A and of B between LDW/HDW and the HDW-enriched zone. It is labelled ‘less enriched HDW.’ Water, however, retains enough enrichment of HDW and high enough concentrations of H+ and OH– to catalyze the reaction, producing C + D.
Although C and D have greater affinity for HDW than have A and B (or the reaction would not take place), their concentrations in the larger volume of LDW/HDW are both zero, allowing them to diffuse out spontaneously.
To summarize: each time a solute is taken up into HDW:
- folding of the parent protein loosens, as HDW increases, allowing the cleft to open to the influx of water.
- LDW/HDW moves into the zone of HDW surrounding the solutes
- where it is in the zone of positive pressure, it is converted to HDW
- the extra water cannot all fit in the zone of positive pressure: it encroaches on the zone of negative pressure (compare Figures 2a and b)
- the average enrichment of HDW in the zone nearest the surface is lower than it was initially.
- concentrations of all solutes in HDW decrease because their partition coefficients are lowered.
Following loss of A, B, C and D, the protein reverts to its original folded state.
Apparent Saturation Kinetics
As the concentration of A (or B) in the outside solution increases, its partition coefficient decreases (see above). Therefore, the increase in rate with increasing concentration levels off, which gives the appearance of a saturated binding site and Michaelis-Menten kinetics.
Promoters and Inhibitors (Allostery)
Any molecule, which partitions into HDW, partially or totally inhibits this reaction by decreasing enrichment of HDW in water surrounding the reactants. Thus a higher external concentration is needed to obtain the same rate of reaction. On the other hand, a molecule which partitions into LDW induces more LDW, compacting the enzyme and preventing swelling, so that there is no decrease in the partition coefficients. In fact partition coefficients increase because the inability of water to move in to abolish the osmotic pressure gradient acts as a pressure and increases the enrichment of HDW surrounding solutes (see under cellulose acetate).
The Same Reaction Catalysed by an Enzyme that Partitions into LDW
This enzyme folds to reduce excess HDW that is induced by its elongated state. When A and B are taken up selectively into HDW, the compensatory flux of water must induce more HDW. Thus the enzyme folds more tightly, somewhat closing the cleft and the flux of water is not permitted. Therefore the osmotic pressure gradient stands, and acts as an additional pressure on all enriched HDW, increasing its enrichment The reaction takes place extremely rapidly in this highly enriched HDW with increased concentrations of H+ and OH–. Products, presumably, can still leave to their zero external concentrations.
A Representative Reaction in LDW
Let the enzyme be one that partitions into HDW and induces LDW, and let the reaction be
ADP + KPi = ATP
2. The reaction takes place with spontaneous production of ATP.
ADP2- and ATP3- and K+ have extremely large partition coefficients between LDW and LDW/HDW. This is entirely because of their ionic character. Anions in general, and phosphates in particular, have very great affinity for LDW, as has K+. They, therefore, create a steep osmotic pressure gradient. If water moves in to abolish this gradient, it will move into the region under negative pressure, thus converting much HDW to LDW. But this enzyme has folded in order to decrease its excess production of LDW. Therefore induction of more LDW and opening up of the cleft is prohibited. The osmotic pressure gradient, therefore, stands.
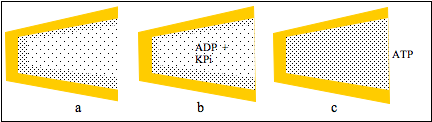
Figure 3: a cleft between protein domains contains zones of HDW and ofLDW; b, ADP and KPi partition into LDW which is not permitted to swell; thereaction takes place and c, the pressure due to the uncompensated osmoticpressure gradient converted LDW to HDW so that ATP was released.
Figures 3a and b show that uptakes of ADP and KPi do not increase LDW and do not open the cleft. The reaction takes place but the inability of water to move becomes a pressure acting on all the water in the cleft. In Fig.3c it converts LDW to HDW and releases all solutes from LDW, including ATP. Following release of solutes, zones of HDW and LDW reform at the surfaces (Fig 3a). This action of K+ salts to convert LDW to HDW with release of all solutes was observed in neutral cellulose acetate films with narrow pores. (See below.)
Cellulose Acetate Membranes
The infra red spectra of water in cellulose acetate membranes confirmed the basic hypothesis on which this treatment of enzymes rests (Wiggins and van Ryn, 1986). The membranes were soaked in water or solution; blotted dry; and their water contents, OH-stretch difference, infrared spectra, and ion contents measured. They did not swell: presumably the cross-linked matrix was too rigid. The three peak wave numbers of interest were: 3250 cm-1 (the value in ice, and, presumably LDW; 3444 cm-1 (bulk liquid water) and 3635 cm-1 (presumably HDW). Membranes soaked in water had a large peak at 3250 cm-1 and a smaller peak at 3635 cm-1 but nothing at 3444 cm-1.
Membranes soaked in 100 mM LiCl had a greatly increased peak at 3635 cm-1 which was as large as the peak at 3250 cm-1
Membranes soaked in 10 mM KCl had a broad band with a single peak at 3444 cm-1. These are now interpreted in the following way:
· the two peaks that revealed themselves inside the membrane pores, inexplicable at the time, are the overlapping bands in liquid water that have since been proposed to account for the single broad band at 3444 cm-1 (Wiggins, 2009).
· 100 mM LiCl accumulated into HDW, creating an osmotic pressure gradient which could not be eliminated by flux of water. It acted instead as a pressure gradient, greatly increasing the enrichment of HDW. The LiCl concentration in the membrane water was 22 mmol/kg water; i.e., although LiCl was accumulated into HDW, it was strongly excluded from LDW.
· 10 mM KCl accumulated into LDW, creating an osmotic pressure gradient which could not be eliminated by flux of water. It acted as a pressure on the water in the pore converting it to HDW with loss of all KCl. Immediately afterwards zones of HDW and LDW reformed in the pores. Thus, while bulk water was a mixture in space of microdomains of LDW and of HDW, pore water in the presence of 10 mM KCl, oscillated in time between HDW and LDW. They had the same single broad spectral band at 3444 cm-1. The observed concentration of KCl in the membranes was 10 mmol/kg water: i.e., it oscillated between 0 mM (in HDW) and some undetermined high concentration (in LDW).
Small-pored polyamide beads also reached a limit in swelling. They selectively took up a molecule like glucose with a modest partition coefficient, but then released it rapidly upon addition of 100 mM KNO3 which, evidently, created an osmotic pressure gradient requiring a larger flux of water than the matrix would allow. In the enzyme the limiting factor was not rigidity of the matrix but the force folding the protein to its active conformation, which in the present case was elimination of excess LDW. This has been suggested as the mechanism of sodium channel-opening (Wiggins, 2007). If water in the entrance compartment of the channel is predominantly LDW, the channel is closed to Na+. If K+, then accumulates into the LDW and water is not permitted to follow it to eliminate the osmotic pressure gradient, LDW converts to HDW and the channel loses K+ and is open to Na+. After passage of Na+ water reverts to LDW.
Reaction in LDW with Swelling
Let the enzyme be one that has partitioned into LDW and induced HDW. Let the reaction be
Uptake of X and Y into LDW produces more LDW, which loosens the folding of the protein and allows water to move in and abolish the osmotic pressure gradient. Most extra water goes into the zone under negative pressure, but, as with the corresponding reaction in HDW, some extra water encroaches on the region of positive pressure, or it moves out beyond the zone in which the negative pressure operates. In either case it loses some of its initial enrichment in LDW. The reaction takes place and Z diffuses out into the larger volume of LDW/HDW in which its concentration is zero.
This reaction also has saturation kinetics and is promoted by molecules which partition into HDW and inhibited by molecules that partition into LDW.
Reaction at a Charged Site
Each charged group on the surface of an enzyme generates pockets of enriched HDW and LDW because the counterion creates an osmotic pressure gradient which acts as a pressure gradient. Water in the compartment marked out by the counterion, diffusing under the influence of the fixed charge, becomes enriched in HDW, while an adjacent zone under negative pressure becomes enriched in LDW. The degree of enrichment of both zones depends upon the nature of the counterion, and the presence of other solutes. Figure 4 illustrates a typical negatively charged site with Na+ as counterion. The pressure gradient is shown in red.
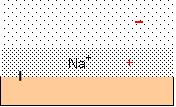
Figure 4: a negatively charged surface has Na+ ascounterion. The osmotic pressure gradient acts as a pressure gradient, inducingHDW in the pocket defined by the diffusion of Na+ and .LDW furtherout. The pressure gradient is shown in red.
Dextran sulphate, a highly charged polymer, has been used to determine the effects of added solutes upon the viscosity of its solutions.
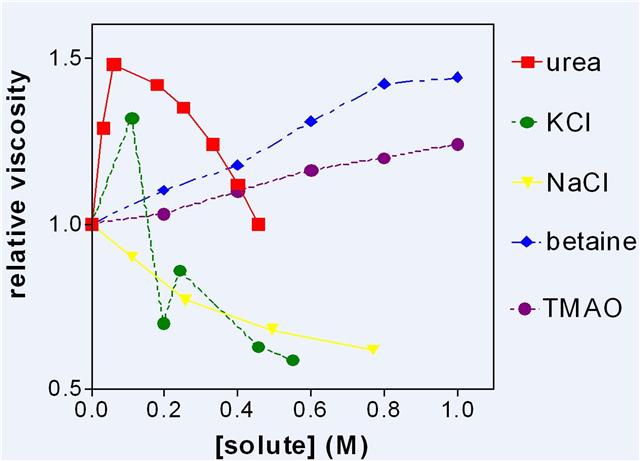
Figure 5: The viscosity of dextran sulphate solutions (3 g water to 1 g sodium dextran sulphate) to which various solutes were added.
Figure 5 shows some representative results which were reproduced many times. It was assumed that increase in viscosity was due to increase of LDW and/or decrease of HDW. Urea which partitions into LDW is of particular interest, as it appears to be one of the more eccentric molecules in Figure 5 and it links the discussion of enzymes to the properties of charged surfaces. Low concentrations of urea increased LDW by partitioning into it and drawing other water into the same zone of negative pressure either from the external solution of LDW/HDW or from the neighbouring zone of HDW. It then quite suddenly stopped increasing LDW at about 0.1 M and viscosity steadily decreased with higher concentrations. From these results alone, it is not possible to determine the source of the water that abolishes the osmotic pressure gradient caused by selective uptake of urea. By combining this result with that of other experiments, however, the decision could be made. It became clear that below 0.1 M, urea drew water from the counterion zone and that above 0.1 M it had exhausted that source of water and thereafter the main effect was a decrease in viscosity as urea increased HDW in the rest of the solution. The extreme sharpness of this transition is comparable with that shown by 10 mM KCl in cellulose acetate membranes and, indeed by KCl in Figure 5, suggesting that urea and KCl both exhaust available HDW at about 0.1 M, convert water to HDW, and lose all solutes.
The other experiments which made this conclusion possible consisted of measuring the rate of loss of silicates from the surfaces of small glass beads or of charged groups from cation or anion exchange resins. Urea below 0.1 M greatly accelerated these losses. Cleavage of silicates from the surface only takes place in highly enriched HDW in which concentrations of H+ and OH–are both high. This shows that in the presence of urea or KCl, water was leaving the HDW zone for the LDW zone so that the concentration of the counterion increased and the enrichment of HDW increased. This is just one example of the general principle that measurement of a single property of a system does not give enough information for a uniquesolution. Another example from the same pair of experiments is that NaCl, which decreased the viscosity of dextran sulphate solutions, protected silicates from cleavage by HDW. NaCl partitions into HDW, increasing the dielectric constant of the solution round the fixed charge, and allowing water to move in so that there is an increase in the amount of enriched HDW but not in its degree of enrichment. These apparently simple systems are in fact complex.
Dangers of Charged Sites
Enzymes must beware of charged sites because there are many ways in which HDW becomes reactive enough to cleave the bond linking the charged group to the backbone. Loss of an essential amino acid residue or cleavage of a peptide bond would destroy the enzyme. Mg2+ as counterion can often induce this level of reactivity. Electrophoresis of DNA with Tris as counterion gave a series of bands showing that many different sized oligonucleotides were present. When Mg2+ was counterion, the bands all disappeared. Presumably the oligonucleotides that had been identified with Tris had been hydrolyzed by highly reactive HDW. Since Mg2+ is divalent, it was held very closely to the charged group, which was therefore contained in a small volume of highly enriched HDW. The osmotic pressure gradient increased further because, in order to maintain electroneutrality, the divalent counterion was accompanied by an anion. The extreme effects of Mg2+ as counterion are obscured by the presence of excess MgCl2, which behaves like NaCl in Figure 5. Again two sets of experiments are needed to understand its action. It has been shown that the cation transport ATPases apparently make subtle use of the power of Mg2+, at the same time evading the danger.
MgATP can enter most zones of water because it comprises a chelate of Mg2+, which partitions strongly into HDW, and ATP, which partitions strongly into LDW. Its own partition coefficient is therefore quite low. In the Na,K-ATPase, for example, it phosphorylates an aspartyl residue in the cleft of the active site, leaving Mg2+ as the counterion. Since there is only a single Mg2+, its effect is not opposed by excess MgCl2, and it readily out-competes other cations at higher concentrations because it has the greatest affinity for HDW and is divalent. The result is an extremely enriched pocket of LDW to accomplish the transport step and an extremely enriched pocket of HDW to hydrolyze the phosphoenzyme. (Wiggins, 2007). Na+ is pushed up to the apex of the cleft by an advancing wave of LDW and out through an open channel, while K+, with its high affinity for LDW, moves in. K+, more powerfully than urea, accumulates into LDW, extracting water from the region of positive pressure and completing the enrichment of HDW, which then hydrolyses the phosphoenzyme. In this way the enzyme uses the unique properties of water at a charged site with Mg2+ as counterion, but retains its integrity. Many enzymes are phosphorylated, used, and dephosphorylated in this way.
Oxygenation of Hemoglobin
Oxygenation of hemoglobin (Matthews and van Holde, 1990) may be a process that can be simply explained in terms of LDW and HDW. Four chains fold to achieve the active state. The reaction takes place in the heme which has a Fe atom held by bonds donated by nitrogens in the heme. This Fe is doubly positively charged with two counterions that generate their own localized pockets of highly enriched HDW and highly enriched LDW. It is a powerful system because with two counterions it generates double the usual osmotic pressure gradient.
This results in differences from reactions so far discussed:
· the volume of the compartment under pressure is determined by the diffusive path marked out by the counterions, which are assumed to be Cl– -the most common anion in the extracellular solution.
· the volume increases only if the local dielectric constant increases with uptake of electrolytes (eg NaCl) which partition into HDW: as the concentration of NaCl increases, the volume of the compartment increases and allows influx of water to abolish the osmotic pressure gradient.
· The volume decreases with uptake of a solute which decreases the dielectric constant (e.g., a hydrophobic solute).
· The volume also decreases if a solute partitions selectively into LDW, drawing water from the counterion compartment, a movement which is permitted because it reinforces (rather than opposes) the electrostatic force
· All movements of water are internal exchanges so that they depend, not upon the folding of the protein, but on the control of the electrostatic field, which generates much stronger forces than those of osmotic systems.
Hemoglobin
There are several observations that have to be addressed: oxygen must be taken up strongly to be transported round the body, but it must also be released in tissues where it is needed.
1. Cooperative Binding of Oxygen
Oxygen binds weakly at low concentrations but the binding strength increases with its concentration. In terms of LDW/HDW, oxygen partitions into enriched HDW in the pocket occupied by counterions. Water does not follow to abolish the osmotic pressure gradient. As the concentration of oxygen increases, the standing osmotic pressure gradient increases, the pressure gradient increases and water is increasingly enriched in HDW. This accounts for ‘weak binding’ at low concentrations and ‘strong binding’ at high concentrations.
2. Conformational Change When Oxygen Binds
Although folding of the enzyme does not control movement of water at the charged site, production of either HDW or LDW during the reaction must generate a change in folding. Accumulation of oxygen in HDW increases enrichment of HDW in that pocket. This appears to result in a tightening of the folded structure because X-ray crystallography shows that a central hole decreases when oxygen is taken up. Presumably, the extended hemoglobin chains induced more HDW on solution in LDW/HDW (Wiggins, 2009).
CO2 and Biphosphoglycerate
These two compounds modify oxygen uptake. CO2 at physiological pH is principally HCO3–. Like most anions (including biphosphoglycerate) HCO3– partitions into LDW, taking a cation with it. As it does so it draws water from the zone under positive pressure, decreasing the volume of HDW accessible for uptake of O2 and slightly increasing the standing osmotic pressure gradient and the degree of enrichment in HDW. The net result appears to be that uptake of oxygen is slightly decreased. This proposal can be tested experimentally in, perhaps, a dextran sulphate solution.
The Bohr Effect
At extremely low concentrations of venous O2 there is a drop in pH. Of univalent cations, H+ ions have by far the greatest affinity for HDW. Even from low concentrations they are taken up, together with an anion, probably Cl–, increasing the dielectric constant in the zone under pressure, allowing entry of water from the associated zone of LDW. This dilutes not only oxygen, but also the two counterions which determine the osmotic pressure gradient, so that both concentration of oxygen and enrichment of HDW are depressed, with further loss of oxygen.
This scheme also allows ‘binding’ of O2 and HCO3– at the same site.
Role of Aminoacid Sequences
The infra red spectra have confirmed that pores in membranes or clefts in proteins contain both LDW and HDW. If, as seems probable, HDW zones are immediately close to the surface, the amino acid sequences in the active site become another factor to consider. The surface of a protein is, in fact, an array of side chains with some kinetic energy. The HDW zone will allow hydrophobic side chains to extend freely from the polypeptide chain, but side-chains which have an affinity for LDW tend to lie flat along the surface. Thus the local composition of the polypeptide chain controls to a degree the volume available to the reaction.
Conclusion
The long neglected variable, pressure (Franzese et al, 2008), makes this scheme versatile and, perhaps, credible.
References
Franks F (1981). Polywater. Cambridge Mass MIT Press.
Franzese G, Stokely K, Chu X-Q, Kimar P, Mazza MG, Chen S-H and Stanley HE (2008). Pressure Effects in Supercooled Water, J. Phys. A Condensed Matter 20: 494210.
Matthews CK, van Holde KE (1990). Biochemistry. The Benjamin/Cummings Publishing Company, New York, 373.
Wiggins PM and van Ryn RT (1986). The solvent properties of water in desalination membranes. J Macromol Sci-Chem A23: 875-903.
Wiggins PM (1988). Water structure in polymer membranes. Progress in Polymer Science 13: 1-35.
Wiggins PM (2002). Enzymes and two-state water. J. Biol. Phys Chem. 2: 25-37.
Wiggins PM (2007). Life depends upon two types of water. PloS ONE 3 (1): e1406.
Wiggins PM, (2008a). Prions, plaques and polyelectrolytes. J Biol Phys Chem. 8: 49-54
Wiggins PM (2008b). DNA as a Darwinian self-replicator J Biol Phys Chem. 8: 89-93
Wiggins PM (2009). The Source of Some of the Extraordinary Powers and Properties of Enzymes, WATER 1: 35-41.
Discussion with Reviewers
Frank Mayer1: What about possible influences of water moieties further away from the enzymes? After all, data from a number of workers (e.g., Ling, Pollack) allow to state that a distribution of LDW and HDW moieties may exist that may be different from that depicted in the figures of the article (“exclusion zones” may be of importance).
Philippa Wiggins: I intended this article to be about enzyme reactions which must take place within a very few nm of the surface. Exclusion zones and more distant interactions of water molecules (a la Ling) are not excluded but are certainly not included in this limited treatment. They are just irrelevant. The particular mechanism that I have proposed for induction of HDW and LDW at surfaces is extremely unlikely to extend far into the solution. Experiments with polyamide beads have suggested that a diameter of 3 nm is about maximum. That means 1.5 nm at a planar surface.
Mayer: Many data are available which indicate that membrane-associated enzymes are not directly attached to the surface of the membrane, but connected to the membrane by “stalks” 3 to 7 nm long, or that typical “soluble” enzymes are, in fact, also connected to membrane surfaces by stalks of this size. Could that mean that a size range between 3 and 7 nm is the optimum for the positioning of the active site of an enzyme relative to a surface (the membrane surface) that is known to influence water structure?
Wiggins: I suggest that these stalks are devices to protect the membrane not to pander to enzymes. If an enzyme that partitioned into HDW diffused all the way to the membrane it would meet a barrage of charged headgroups and counterions: phosphates with Ca2+ and amino groups with Cl–. It would then diffuse into a pocket of HDW and unfold. It originally folded only because it had to decrease the excess LDW that it induced in its extended state. In the pocket of HDW, however, it is under positive pressure and cannot induce LDW. Therefore there is no driving force for it to fold. In its elongated state it greatly reduces the dielectric constant at the fixed charge so that the counterion moves in closer to the fixed charge. This increases the osmotic pressure gradient, the pressure gradient and the degree of enrichment with HDW. It cleaves the phosphate or the amino group off the membrane, destroying its barrier properties. I have suggested (Wiggins, 2008a) that prions wreak their havoc by this means. Your distance of 3 to 7 nm is to be expected for a charged site. The 2-3 nm is the thickness at an uncharged surface. The only paper I have read that measured the thickness of the double layer gave a value of 6 nm. It depends upon the counterion. This measurement was made with Zn2+.
Mayer: Could it be envisaged that the structural state of the water at a distance of about 3 to 7 nm from the enzyme cleft sets the basic conditions, and that reactions/alterations of water structure taking place within the cleft are “embedded” into (or even regulated by) the basic conditions?
Wiggins: You would have to come up with a good mechanism. There may well be one. I am particularly inclined toward a 2-3 nm limit because the original division of water into microdomains involves a decrease in entropy which can only increase as the size of the domains increases. According to Gene Stanley, the microdomains separate into two solutions at about -50oC. This would involve a very large decrease in entropy.
Ivan Cameron2: My question concerns the hemodynamics on oxygenation of hemoglobin. It seems likely that erythrocyte shape change and shear force (stenosis) occurs when erythrocytes pass through the capillary bed. This perturbation probably disrupts their LDW state and thereby modifies oxygen as well as CO2 exchange. What is your comment on this possibility?
Wiggins: This pressure acts on the whole red cell. It may well modify the C02/O2 exchange, but it would be rapidly reversible. But that is a very good point.
Cameron: How can your water model of enzyme action be tested?
Wiggins: There is a wealth of experimental data on enzyme reactions, all interpreted in terms of binding sites. If these can also be interpreted in terms of surface water, then that is a good test of the mechanism. I have done that here with Michaelis-Menten kinetics and with allostery. There are many more things one could do, but my personal ignorance of biochemistry gets in the way. As I said earlier, there are plenty experiments showing the properties of water at other surfaces, and no reason why protein surfaces should be different.
1 Head, Structural Biology Department, Georg-August-University, Göttingen, Germany.
2 University of Texas Health Science Center at San Antonio, Graduate School of Biomedical Sciences, Cellular and Structural Biology.