Experimental Determination of Proton-Cation Exchange Equilibrium Constants at Water-Membrane Interface Fundamental to Bioenergetics
Experimental Determination of Proton-Cation Exchange Equilibrium Constants at Water-Membrane Interface Fundamental to Bioenergetics
Haitham A. Saeed and James W. Lee*
Department of Chemistry and Biochemistry, Old Dominion University, Physical Sciences Building 3100, 4541 Hampton Blvd, Norfolk, VA 23529
*Corresponding Author email: jwlee@odu.edu
Keywords: water thixotropy, viscosity, conductivity, ions, hydrophilic surfaces, laser light scattering, luminescence, ultraviolet absorption, proton-electrostatic localization model, cation exchange equilibrium constant, corrected pmf equation, salinity tolerance, bioenergetics, proton conductor.
Received: October 11, 2017; Revised: March 16, 2018; Accepted: March 23, 2018; Published: May 2, 2018; Available Online: May 2, 2018
Abstract
Recently the Lee proton electrostatic localization hypothesis has successfully elucidated the decades-longstanding energetic conundrum of ATP synthesis in alkalophilic bacteria. According to the Lee proton electrostatic localization model, the equilibrium constant KPi for non-proton cations such as Na+ to delocalize the localized protons from the membrane-water interface should be much smaller than unity. Through the experimental study reported here, it has now been determined for the first time that the equilibrium constant KPi is indeed far much less than one. The equilibrium constant KPNa +for sodium (Na+) cations to exchange with the electrostatically localized protons was determined to be (5.07 ± 0.46) x 10-8 while the equilibrium constant KPK+ for potassium (K+) cations to exchange with localized protons was determined to be (6.93 ± 0.91) x 10-8. These results mean that the localized protons at the water-membrane interface are so stable that it requires ten million more sodium (or potassium) cations than protons in the bulk liquid phase to even partially delocalize them at the water-membrane interface. This provides a logical experimental support of the proton electrostatic localization theory. The finding reported here may have fundamental implications in understanding the importance of water to life not only as a solvent and substrate but also as a proton conductor for proton coupling energy transduction. It may also have fundamental implications in understanding the salinity tolerance in biological systems in relation to localized proton coupling bioenergetics.
Abbreviations and Acronyms
| pmf (∆p) | Proton motive force |
| ∆μH+ | Proton electrochemical gradient |
| ∆ψ | The trans-membrane potential generated due to the difference in electric potential across the biological membrane |
| ∆pH | The pH difference between the two bulk aqueous phases separated by the membrane |
| ε | Dielectric permittivity |
| pHnB | Stroma (or cytoplasmic) bulk phase pH |
| pHpB | Lumen (or periplasmic) bulk phase pH |
| [HL+] | Effective concentration of the localized protons at the membrane-water interface at equilibrium with non-proton cations |
| [HL+]0 | Effective localized proton concentration at the membrane-water interface without cation exchange. |
| C/S | Membrane capacitance per unit surface area |
| ĸ | Dielectric constant of the membrane |
| d | Thickness of the membrane |
| l | Thickness of the localized proton layer |
| ĸpi | Equilibrium constant for non-proton cations to exchange with the localized protons at the water-membrane interface. |
| [M i+pB] | Concentration of the non-proton cations in the bulk phase of the liquid culture medium. |
| [H+pB] | Concentration of protons in the bulk phase of the liquid culture medium |
| ∆G | Gibbs energy change |
| PI | Proton sensitive membrane interface site facing the anode (P) water chamber. |
| NI | Proton sensitive membrane interface site facing the cathode (N) water chamber. |
| PB | Proton-sensitive film applied in the middle of the anode chamber water bulk phase |
| NB | Proton-sensitive film applied in the middle of the cathode chamber water bulk phase |
| CB | Proton-sensitive film placed into the bulk liquid phase of the Teflon center chamber |
| P′ | Proton-sensing film placed at cathode site facing the solution within the Teflon center chamber |
| N′ | Proton-sensing film placed at anode site facing the solution within the Teflon center chamber |
| rD | The Debye length |
| [NaL+] | Localized sodium ions concentration on the water-membrane interface |
| KPNa+ | Equilibrium constant for Na+ exchange with localized protons |
| KPK+ | Equilibrium constant for K+ exchange with localized protons |
Introduction
Peter Mitchell’s work on chemiosmotic theory (1-3) and its central bioenergetics equation has been incorporated into many textbooks (4-6). In one of its forms, this equation is expressed as the proton motive force (pmf) across a biological membrane that drives protons through the ATP synthase:
pmf =∆ψ + (2.3 RT) ∆pH/F [1]
where ∆ψ is the electric potential difference across the membrane, R is the gas constant, T is the absolute temperature, and ∆pH is the pH difference between the two bulk aqueous phases separated by the membrane. In this framework, the protons are considered to be solutes, similar to sugar molecules, that are delocalized, existing everywhere in the bulk aqueous phases. Consequently, the Mitchellian view of bioenergetics is that the ATP synthase is coupled to the redox proton pumps via bulk phase-to-bulk phase proton electrochemical potential gradients generated across the biological membrane. The chemiosmotic theory was a major milestone in the history of bioenergetics; its significance to the field could hardly be overstated.
However, the question as to what extent the proton coupling pathway for producing ATP is delocalized throughout the bulk aqueous volume or localized at the membrane surface has remained under discussion since it was first raised in 1961 by Williams (7-11). Perhaps the most well-established observations that disagree with the Mitchellian equation [1] are in alkalophilic bacteria, such as Bacillus pseuodofirmus (12-14). These bacteria keep their internal pH about 2.3 units more acidic than the ambient bulk pH of 10.5, while its membrane potential is about 180 mV (15-17). The use of the Mitchellian equation [1] in this case would yield a pmf value so small (44 mV at T = 298K) that it has remained a mystery for the last three decades as to how these organisms are able to synthesize ATP (18-20).
Recently, Lee has put forward the proton electrostatic localization hypothesis for a natural mechanism to produce surface membrane localized protons (21, 22), which is built on the premise that a water body acts as a proton conductor. This premise is consistent with the well-established knowledge that protons quickly transfer among water molecules by the “hops and turns” mechanism first outlined by
Grotthuss two centuries ago (23-25). Considering a conceptualized cell system consisting of an impermeable membrane immersed in pure water with an excess number of free protons (H+) outside and an equal number of free hydroxyl ions (OH–) inside, and given that pure water acts as a proton conductor, it follows mathematically from applying the Gauss Law of electrostatics that the excess protons and anions are respectively localized at each of the two water-membrane interfaces along the two opposite sides of the membrane, forming a protons-membrane-anions capacitor-like structure (22). For an idealized proton capacitor, the concentration of the localized protons [HL+]O at the membrane-water interface is related to the membrane electric potential difference by
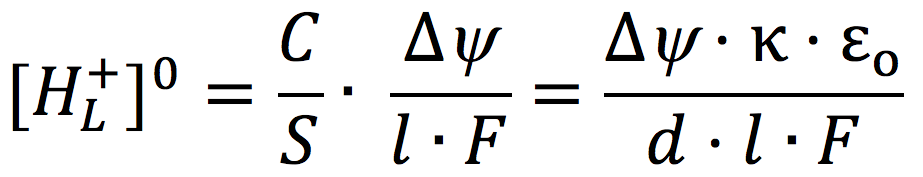
[2]
where C/S is the specific membrane capacitance per unit surface area, l is the thickness of the localized proton layer, κ is the dielectric constant of the membrane, ε0 is the electric permittivity, and d is the membrane thickness.
Considering a biological cell system, it is important to note that non-proton cations in the aqueous media may exchange with protons localized at the membrane surface and thereby reduce their concentration. According to the Lee proton electrostatic localization hypothesis (22), such exchange effects can be expressed by augmenting equation [2] for the concentration of surface localized protons as
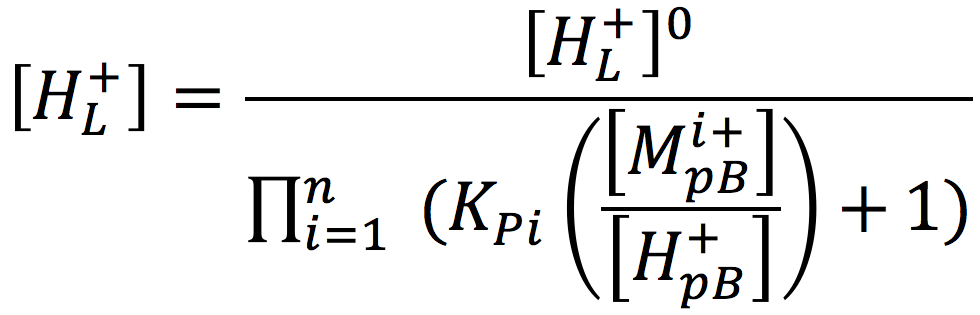
[3]
Where [H+pB] is the proton concentration in the bulk aqueous phase, [Mi+pB] is the concentration of non-proton cations, and KPi is the equilibrium constant for non-proton cations to exchange with the localized protons. Thus, it is to be expected that the non-proton cation concentrations that occur in biological systems may play a significant role in modulating the proton motive force for varieties of biological functions including the production of ATP.
Furthermore, according to the Lee proton electrostatic localization hypothesis (22), the textbook (4-6) Mitchellian classic pmf equation [1] shall now be revised by combining the concentration of surface localized protons [HL+] with the concentration of bulk phase protons, explicitly:
![]()
[4]
Here, as in equation [3], [H+pB] is the proton concentration in the periplasmic bulk aqueous phase while [H+nB] is the proton concentration in the cytoplasmic bulk phase. As discussed in the Lee proton electrostatic localization model (22), this newly formulated pmf equation with its definition of localized protons [HL+ ] as shown in equations [2] and [3] also adds clarification on the origin of electric potential difference term ∆ψ well beyond the Mitchellian equation [1] that contains the term ∆ψ but does not seem clearly explain its origin. In Lee’s newly formulated pmf equation [4], the electric potential difference term ∆ψ that helps to drive protons through the ATP synthase is also the same factor in equation [2] that determines the concentration of surface localized protons which are available to the ATP synthase. Indeed, according to equation [2], ∆ψ exists precisely because of the excess cations (including H+) and the excess anions (such as OH–) charge layers localized on the two sides of the membrane in a protons-membrane-anions capacitor-like structure (22). This explains the origin of ∆ψ and its relation with the concentration of localized protons [HL+ ] as expressed in equations [2] and [3]. Moreover, it is expected that the surface localized protons would make a very significant contribution to the protons available for driving ATP synthesis. In fact, applying equation [4] for the first time to the alkalophilic bacteria case noted above, using some preliminary estimates for the quantities in equations [2] and [3], recently has already yielded a pmf value of ~225 mV, which is 5 times larger than the value obtained from equation [1] and is sufficient to overcome the observed phosphorylation potential in order to synthesize ATP (22).
Biological membranes are made of phospholipids which have negatively-charged phosphate groups. The presence of a negative charge on the biological membrane surfaces produce an electric potential that attracts the counter-ions (of opposite charges) and repels ions carrying the same charge as that of the surface. Several theories have been introduced which attempt to determine the surface potential of charged surfaces and to describe the electrical phenomena at the surfaces of the biological membranes (26, 27). The diffuse electric double-layer presented by Gouy-Chapman is currently the model being used for describing the ionic atmosphere near a charged surface whose thickness is estimated as the Debye length (rD). The magnitude of the Debye length — which appears as the characteristic decay length of the surface potential — depends only on the properties of the solution not on the properties of the charged surface (28). For example, a monovalent electrolyte like NaCl solution at 25º C, the Debye length is 30.4 nm at 10-4 M, 0.96 nm at 0.1 M and 0.3 nm at 1 M. This means that the Debye length decreases by increasing the concentration of the electrolytes (29).
Unfortunately, the Debye length cannot be used to estimate the thickness of the localized excess protons because the equations used in calculating the Debye length can be applied only to charge-balanced solutions including 1:1 electrolyte solutions such as NaCl, 2:1 electrolytes such as CaCl2 and 1:2 electrolytes such as Na2SO4 (30). That is, the Debye length equations cannot be applied to estimate the thickness of the localized excess protons layer that does not have counter ions.
Although the Lee proton electrostatic theoretical model (22) has a characteristic localized proton coupling feature, it does not necessarily contradict the electric double layer theoretical model (31). These two models represent two different processes: the former describes the proton motive force with electrostatically localized excess proton coupling bioenergetics while the later belongs to the classic electric double layer phenomenon. For example, the negatively-charged phosphate groups of the biological membrane could attract protons and other cations to its surface forming an electric double layer along the membrane negatively charged surface as expected by the Gouy-Chapman theory (31). However, this double layer always exists at all times during light and dark conditions even when the proton motive force (pmf) is zero. This means that the protons and/or cations attracted to the membrane surface’s fixed charge forming the double layer could not contribute to the proton motive force that drives the flow of protons across the membrane (32).
As illustrated in Figure 1, our previous experimental work demonstrated the formation of a localized layer of excess protons at the water-membrane interface in an anode water-membrane-water cathode system (33), where excess protons were generated by water electrolysis in an anode electrode chamber and excess hydroxyl anions were created in a cathode chamber. When a positive voltage is applied to the anode electrode in water, it first attracts the hydroxyl anions to the anode electrode surface and then counter-ions (protons) distribute themselves near the anions layer, forming a typical “electric double layer” on the anode surface (Figure 1a, right side). When a significant number of excess protons are produced by water electrolysis in the anode chamber (mimicking a biological proton production process such as the respiratory proton pumping system or the photosynthetic water-splitting process), the excess protons electrostatically distribute themselves at the water-surface (including the membrane surface) interface around the water body including a part of the “electric double layer” at the anode surface. From here, it can be seen that the excess proton layer at the water-membrane interface is apparently extended away from the secondary (proton) layer of the “electric double layer” at the anode. The excess proton layer at the water-membrane interface attracts electrostatically the excess hydroxyl anions in the cathode chamber at the other side of the membrane, forming an “excess anions-membrane-excess proton” capacitor-like structure.
Since the membrane is just an insulator layer (not an electrode), the excess proton layer at the water-membrane interface is likely to be a special monolayer (with a thickness probably of about 1 nm), but definitely not an “electric double layer” as that of a typical electrode. The conclusion that there is an excess proton monolayer is also consistent with the known “electric double layer” phenomenon since the excess proton layer can be treated as an extension from the second (proton) layer of the anode’s “electric double layer” (Figure 1a, right side) around the proton-conductive water body surface.
When the electrolysis voltage is turned off, the electric polarization at both anode and cathode disappears and so does the “electric double layer,” leaving only the excess proton layer around the anode chamber water body and the similarly formed excess hydroxyl (anions) layer around the cathode chamber water body as illustrated in Figure 1b. The resulting excess anions-membrane-excess proton capacitor (shown in the middle of Figure 1b) may represent a proof-of-principle mimicking an energized biological membrane such as a mitochondrial membrane system at its energized resting state.
The electrostatic localization conceptualization is quite general. It does not depend on biological scales or processes. Therefore, to provide a first proof-of-principle study (33), we have carried out laboratory bench experiments to create excess protons using an electrolysis set-up with cathode and anode water chambers separated by a proton-impermeable membrane.
It was experimentally demonstrated using a proton-sensing film that excess protons do not stay in the water bulk phase; instead they localize at the water-membrane interface in a manner similar to the behavior of excess electrons in a metallic conductor (33). These observations clearly support the proton-electrostatics localization hypothesis (21, 22) which is a significant contribution to understanding the biological energy transduction processes and the distribution of protons across a biological membrane.
In this paper, we report the effect of cations (Na+ and K+) on localized excess protons at the water-membrane interface by measuring the exchange equilibrium constant of Na+ and K+ cations exchanging with the electrostatically localized protons at a series of cations concentrations. The experimental determination of the cation exchange equilibrium constant with the localized protons reported here will provide a logical support for the Lee electrostatic localized proton hypothesis (22) and gain a more fundamental understanding of the effect of non-proton cations on localized proton population density.
Materials and Methods
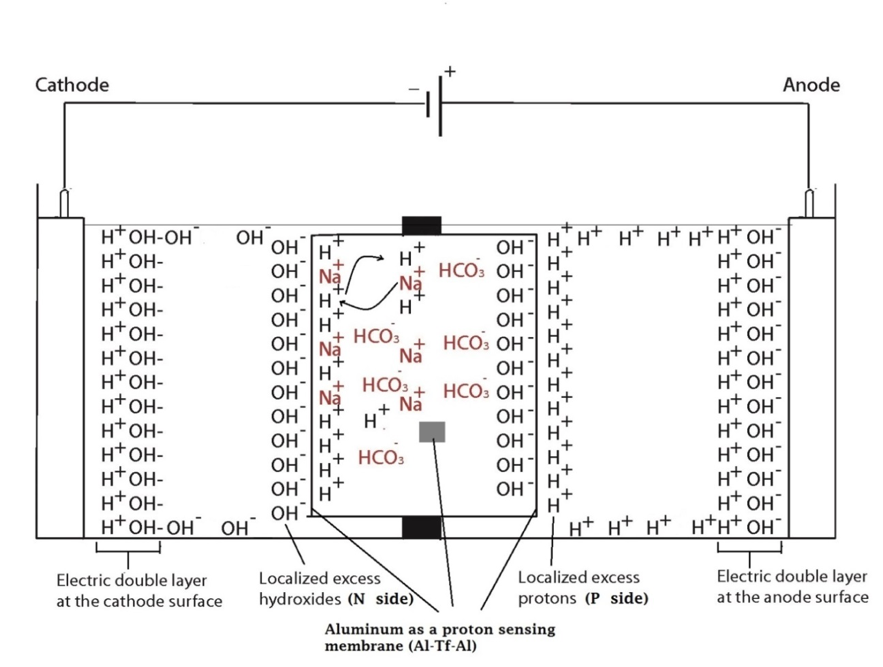
Two ElectroPrep electrolysis systems (Cat no. 741196) purchased from Harvard Apparatus Inc. (Holliston, MA) were used in this experimental study with one of them as a control. Each system comprised a cathode chamber, a small Teflon center chamber and an anode chamber as illustrated in Figure 2. The small Teflon center chamber was inserted to the middle O-ring fitting channel of the inter-chamber wall that separates the cathode and anode water chambers. The ElectroPrep electrolysis system was made of Teflon (polytetrafluoroethylene), a completely inert material that is unreactive under high power voltage.
To test the effect of K+ and/or Na+ salt concentration on localized excess protons, 1.5 ml of pure water or salt solution was placed inside a 1500 µl Teflon center chamber (Harvard Apparatus) as shown in Figure 2. The Teflon center chamber was sealed at each of its two ends by Al-Tf-Al membrane assembly that is formed by sandwiching an impermeable 75-µm thick Teflon (Tf, as known as polytetrafluoroethylene) membrane with two pieces of 25-µm thick proton-sensing aluminum (Al) films (having equal-diameter of 2.35 cm) placed at the two side ends of the Teflon center chamber (internal diameter 1.5 cm and length 0.9 cm). In addition, a small piece of aluminum film was inserted in the middle liquid bulk phase of the Teflon center chamber. The small Teflon center chamber was then placed in between the anode and the cathode compartments of the apparatus as shown in Figure 2 so that one end of the center chamber was in contact with cathode bulk liquid (denoted N side), while the other end was in contact with anode bulk liquid (denoted P side).
The ultrapure MilliQ-deionized water (Millipore, 18.2 MΩ.cm at 22.5 ºC) used in this study was degassed by boiling the water in an autoclave (Yamato Model SM510, Santa Clara, CA) and then it was cooled down to room temperature before use. The two compartments were then filled with degassed ultrapure MilliQ-deionized water (Millipore, 18.2 MΩ.cm at 22.5 ºC): 300 ml in the small compartment where the cathode electrode resides (cathodic compartment) and about 600 ml in the large compartment where the anode electrode resides (anodic compartment), rendering an equal water level in both compartments (Figure 2 shows the schematic diagram of the system and a detailed description of the electrolysis process). This effectively created a “cathode water membrane (Al-Tf-Al) water membrane (Al-Tf-Al) water anode” system.
The pH of the deionized water was measured separately in a small beaker using Orion™ ROSS Ultra™ pH Electrode (Thermo Scientific, Waltham, MA, USA, Cat No. 8102BNUWP) that is designed to measure pH for aqueous samples with very low ionic strength, including ultra-pure water. Two point calibrations were performed using a buffer of pH 7.00, and a buffer of pH 4.01, according to the manufacturer instructions.
A series of experiments with the above settings were performed comparatively in the presence and absence of NaHCO3 or KHCO3 solution for a series of ionic salt concentrations (0, 10, 50, 100, 150, 200 & 400 mM) that was placed into the Teflon center chamber (Figure 2, inset bottom). These experiments were performed to test the effect of sodium or potassium cations on the electrostatically localized protons at the induced P′ side in the center chamber, in comparison with the unperturbed P side facing the anode water. For each experiment, each Teflon center chamber was rinsed with degassed ultrapure MilliQ-deionized water. After sealing one end of the Teflon center chamber with (Al-Tf-Al) membrane, 1.6 ml of degassed liquid water or the ionic salt solution was introduced into the Teflon center chamber through the other end, which was then sealed with another (Al-Tf-Al) membrane. The Teflon center chamber assembly was then fitted into the O-ring channel within the inter-chamber wall that separates the cathode and anode water chambers. The apparatus was then rinsed with ultrapure MilliQ-deionized water to remove any possible salt contamination before loading ultrapure MilliQ-deionized water into cathode chamber (300 ml) and the anode chamber (600 ml). This setup created a “cathode water membrane (Al-Tf-Al) — salt solution — membrane (Al-Tf-Al) water anode” system.
After the apparatus was set up as shown in Figure 2, an electrolysis voltage of 200 V was applied to the system for 10 hours using a digital source-meter system (Keithley instruments series 2400S-903-01 Rev E). To ensure safety, all experiments were performed inside a fume hood that has a built-in air-fan driven ventilation system to disperse the small amount of potentially explosive H2 and O2 gases generated from the water electrolysis process through the fume hood ducts into the outside atmosphere. A second apparatus with the exact same setup and liquid samples except without the 200 V electrolysis voltage was used as a control. The liquid pH was measured of the salt solution inside the Teflon center chamber after the 10-hour period for both the experiment and the control (recording 6 stable pH readings for each sample) using Orion™ ROSS Ultra™ pH Electrode (Thermo Scientific, Waltham, MA, USA, Cat No. 8102BNUWP). Also the conductivity measurement for the water in each of the anode chamber and the cathode chamber at the end of the experiment was performed (recording 6 stable conductivity readings for each sample) using a Beckman coulter conductivity probe (Beckman coulter, Indianapolis, IN, USA, Model 16 x 120 mm, item no. A57201).
Results and Discussion
Demonstration of Electrostatically Localized Protons at the P and P′ Interfaces in a “Water-Membrane-Water-Membrane-Water” System
In the system described above, water in the Electroprep apparatus was electrolyzed at 200 V, forming excess protons / O2 gas in the anode chamber (P) and excess hydroxyl anions / H2 gas in the cathode (N) chamber (Figure 2). Based on the proton-electrostatics localization hypothesis (21), it is predicted that the free excess protons in the anode chamber would migrate and localize themselves primarily at the water-membrane interface (the P site) in the anode (P) chamber. The excess protons localized at the P side would induce an electrostatic localization of hydroxide anions at the other side of the membrane (the N′ site) forming an “excess anions-membrane-excess protons” capacitor-like system (as shown in Figure 3 and on the right of the inset of Figure 2). Similarly, the excess hydroxide anions generated in the cathode chamber would migrate and localize primarily at the water-membrane interface (the N site) in the cathode (N) chamber. It is predicted that this localization of hydroxide ions at the N side would induce electrostatic localization of protons at the other side of the membrane (the P′ site) forming an “excess anions-membrane-excess protons” capacitor-like system (as shown in Figure 3 and on the left of the inset of Figure 2).
These predicted features were indeed demonstrated through observation of localized proton activity on the Al films at the P and P′ sites while there was no observable proton activity at the N and N′ sites, and no observable excess proton activity in the bulk liquid phase at the PB, CB, and NB sites in the three liquid chambers (Figure 4). As predicted by the proton electrostatic localization hypothesis, it was noticed that the proton sensing films (Al-Tf-Al) at the two ends of the Teflon sample chamber showed detection of protons on P and P′ sites adjacent with pure water (in absence of any salt) as shown in Figure 4. The proton-sensing detection employed here was in the form of Al surface corrosion [Equations 5 and 6] when the effective proton concentration was above 0.1 mM (equivalent to a pH value below 4) as explained in Supporting Information, Figure S1 (34, 35). The proton-sensing Al film placed at the N and/or N′ side of the Teflon membrane detected no significant proton activity in that its color remained unchanged (Figure 4).
![]() [5]
[5]
![]() [6]
[6]
It is worth mentioning that the Al film in the Al-Tf-Al membrane does not serve as an electrode since the Al membrane itself was not connected to any external voltage source. It acted as part of a protonic insulating membrane where excess protons accumulated at the surface on the P site while excess hydroxides were localized at the N side as shown in the middle of Figure 3. Therefore, as discussed previously, there was a monolayer of protons localized at the Al membrane surface but there was no double layer formation as expected by Gouy-Chapman double layer theory due to the absence of counter-ions in the anode chamber. However, an electric double layer is expected to be established on the charged electrode surfaces as illustrated in Figure 3. In this three-water-chambers system, we also demonstrated an induced proton layer at the membrane-water interface (the P′ site) in the center liquid chamber with the evidence of proton-sensing Al film at the P′ site showing intense localized proton activity while those at CB and N′ sites showed no protonic activity (Figure 4).
Our bulk-phase pH measurements (Table 1) demonstrated that the Mitchellian proton delocalization view is not true. After the 10-hour water electrolysis, the measured pH value in the anode bulk water body (5.92 ± 0.12) remained nearly the same as that of the cathode bulk water phase (5.81 ± 0.07). If the Mitchellian proton delocalized view is true, there should be significant bulk-phase pH difference (∆pH) between the anode and the cathode water chambers; In contrast, the measured bulk pH data clearly demonstrated again that the Mitchellian proton delocalized view is not true. These bulk water phase pH values averaged from 3 replication experiments (each replication experiment with at least 6 readings of pH measurement in each chamber water, n= 3 x 6 = 18) were statistically also the same as those (5.75± 0.08 and 5.77± 0.21) in the control experiments in absence of the water electrolysis process. Notably, these results again show that excess protons do not stay in the water bulk phase; they localize at the water-membrane interface at the P and P′ sites so that they cannot be detected by the bulk-phase pH measurement as predicted by the Lee proton electrostatic localization model (21, 22).
These are significant results, since both the proton-sensing film detection and bulk liquid pH measurement have now demonstrated, for the first time, that protons can be localized at a water-membrane interface through electrostatic induction at the P′ site in a “cathode water membrane (Al-Tf-Al) water membrane (Al-Tf-Al) water anode” system where the third water body (the center water chamber) is placed in between an anode water chamber and a cathode water chamber interacting in series (Figure 3). The operation of this setup resulted in the formation of two proton capacitors in series: a proton capacitor across the membrane (Al-Tf-Al) with the N and P′ sites and another one across the other membrane (Al-Tf-Al) with the N′ and P sites as illustrated in Figure 2 and Figure 3. This result again shows that liquid water bodies are proton conductors; the behavior of excess protons in proton conductors appears to be similar to that of excess electrons in electric conductors in forming capacitors across insulating membrane barriers.
Equilibrium Constant of Sodium Cation (Na+) in Exchanging with Electrostatically Localized Protons
Demonstration of the localized protons at P′ site in the center chamber enabled us to evaluate the cation exchange of other cations with the localized protons by using salt solutions only in the center chamber without requiring the use of salts in the anode and cathode chambers (Figure 2 and Figure 5). Use of salts in the anode and the cathode chambers which have large volumes would not only cost much more in chemical materials but also might interfere with the electrolysis process complicating the interpretation of the experimental results. Therefore, the utilization of the localized protons demonstrated previously at P′ site with use of salt solutions in the center chamber enabled us to perform quite clean experiments in measuring the effect of other cations on localized protons including determining the cation exchange equilibrium constants with the localized proton population without requiring the use of any salt in the anode or the cathode chambers.
Our experimental results (Table 2) showed that the addition of 10 mM and/or 25 mM sodium ions (sodium bicarbonate solution) in the center chamber had no significant effect on the electrostatically localized protons at the P′ side facing the sodium salt solution, while the use of 75 mM sodium ions (in the center chamber) led to the reduction of electrostatically localized protons populations at the P′ site by about 50%, which was monitored by the color change of the proton-sensing film at the P′ side in comparison with that of the proton-sensing film placed at the positive controls (0 mM sodium ions: water with no salt) P′ site and the P site facing the anode liquid (also no salt). It required the use of 200 mM or higher sodium ion solution in Teflon center chamber to exchange out the localized protons at the P′ site to a level that could not be detected by the proton-sensing Al film (Table 2, row 7). Based on our analysis, this effect of sodium salt (NaHCO3) solution on the localized protons at the P′ site is probably owed to the sodium cations being at higher concentrations (75 mM or above) that may partially exchange with the electrostatically localized protons at the P′ site.
One might reasonably object that the observed effect may be due to the bicarbonate anion and not due to the sodium cation. Therefore, the cation exchange experiments (of different concentrations) were repeated again but with other cations such as K+ which showed the same type of effect on P′ side. But interestingly, in K+ salt solution, the 50% color change at the P′ site was observed at 50 mM (Table 2, row 4) instead of 75 mM. This additional observation further supports that the observed effect was due to cation exchange and not due to a bicarbonate effect. It is well known that the size of potassium cation is bigger than sodium cation. However, in aqueous solution as a free ion, the small sodium ion attracts more water molecules giving it a larger effective diameter compared to the hydrated potassium ion. Since the hydrated radius of potassium ion is smaller than the hydrated radius of sodium ion; its electro-diffusion mobility is faster compared to sodium. It was determined that the mobility of sodium cations under the influence of unit potential gradient (0.53 x 10-3 cm2 V-1 s-1) is slower than potassium cation mobility (0.76 x 10-3 cm2 V-1 s-1) under the influence of unit potential gradient (36). That’s probably why it required a higher concentration of sodium cations (75 mM) to delocalize 50% of the electrostatically localized protons on P′ site and 200 mM Na+ for nearly complete proton delocalization at the P′ site. Moreover, it was reported that fresh solutions of bicarbonate have practically no action on aluminum corrosion (37-40).
The cation exchange equilibrium constant (Kp) can be expressed as:
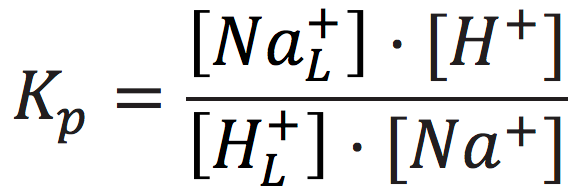
[7]
where [NaL+] is the localized sodium ions concentration at the water-membrane interface (P′ site); [H+] is the concentration of free delocalized protons in the bulk liquid phase; [HL+] is the localized protons concentration at the water-membrane interface (P′ site); and [Na+] is the free sodium ions concentration in the bulk liquid phase.
At the midpoint with 50-50% cation/proton exchange at the localized proton layer, the concentration of the localized non-proton cation would be equal to the concentration of the localized protons. This means that when [NaL+] = [HL+], the cation exchange equilibrium constant (KP) would be
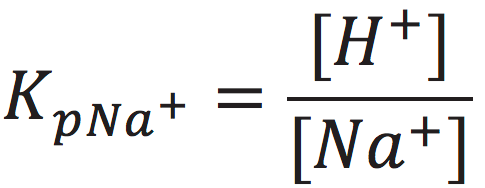
[8]
We observed that the 50-50% cation/proton exchange was achieved when the sodium cation concentration was 75 mM as shown in Table 2 (and Table S7). At 75 mM of sodium cation concentration (the midpoint), the amount of localized protons on the proton sensitive membrane was decreased to half compared to that of the positive control in the absence of sodium cations. The pH of the sodium salt solution (75 mM) inside the Teflon center chamber before the sodium cation/localized proton exchange process was found to be 8.37 ± 0.09 as shown in Table 3 (and Table S17). By using this pH value for the bulk proton concentration [H+] and the known sodium cation concentration (75 mM) in equation [8], the sodium/proton cation exchange equilibrium constant was calculated to be 10-(8.37 ± 0.09) M /0.075 M= (5.86 ± 1.2) x 10-8.
We noticed that the pH of the sodium ion solution (75 mM) was slightly changed during the cation-proton exchange experiment using the Al film-based proton sensor (Equations 5 and 6) at P′ site. The final pH value of the bulk sodium bicarbonate solution after 10 hours experimental run at 200V was 8.48 ± 0.07. Using this final pH value (8.48 ± 0.07) for the bulk proton concentration [H+] and the known sodium cation concentration (75 mM) in equation [7], the KPNa+ value was calculated to be 10-(8.48 ± 0.07) M /0.075 M= (4.45 ± 0.73) x 10-8, which is slightly smaller than that calculated using the initial pH (8.37 ± 0.09). The true KPNa+ value is likely to be in between with an average of (5.07 ± 0.46) x 10-8.
Equilibrium Constant of Potassium Cation (K+) in Exchange with Electrostatically Localized Protons
Similarly, at 50 mM potassium cation concentration (the midpoint), the amount of localized protons on the proton sensitive membrane was decreased to half compared to that of the positive control in the absence of potassium cation (Table 2 and Table S8). The pH of the potassium salt solution (50 mM) inside the Teflon center chamber before the potassium/proton exchange process (Table 4 and Table S18) was determined to be 8.45 ± 0.03. By using this pH value for the bulk proton concentration [H+]
and the known potassium cation concentration (50 mM) in
![]()
the potassium/proton cation exchange equilibrium constant was calculated to be 10-(8.45 ± 0.03) M /0.050 M= (7.20 ± 0.59) x 10-8.
The final pH value of the bulk potassium bicarbonate solution after a 10-hour experimental run at 200V was 8.48 ± 0.13. Using this final pH value (8.48 ± 0.13) for the bulk proton concentration [H+] and the known potassium cation concentration (50 mM) in
![]()
the KPK+ value was calculated to be 10-(8.48 ± 0.13) M /0.050 M= (6.85 ± 1.99) x 10-8, which is slightly smaller than that calculated using the initial pH (8.45 ± 0.03). The true KPK+ value is likely to be in between with an average of (6.93 ± 0.91) x 10-8.
The bulk concentration of potassium cations after the potassium/proton exchange process used in determining the potassium cation exchange equilibrium constant was the concentration that resulted in 50-50% cation/proton exchange at the localized proton layer (i.e.: 50 mM K+). This is because the amount of localized protons that was exchanged out by the potassium cations was likely to be so small that it would not significantly reduce the 50 mM K+ concentration. It has been determined in a previous study (33) that the amount of localized proton density at the PI site that has an effective area 2.55 cm2 to be 1.19 x 10-9 moles/m2 which is equivalent to a localized pH value of 2.92 assuming an 1-nm proton layer thickness. However, after 50-50% cation/proton exchange, 1.52 x 10-13 moles of K+ from the bulk liquid phase would move into the localized proton layer, exchanging out an equal amount of localized protons into the bulk liquid phase. The removal of 1.52 x 10-13 moles of K+ from the bulk salt solution (1.5 ml) would reduce the bulk liquid phase K+ concentration by 1.01 x 10-7 mM, which is negligible compared to the initial K+ concentration (50 mM).
Other Related Observations with Electrostatically Localized Protons
During the experiments, we noticed the importance of using ultrapure Millipore water that does not contain too much dissolved gases. For example, during the winter season when the laboratory temperature (typically about 22ºC) is significantly higher than the outside water supply, the Millipore water supplied from a cold air-saturated water source often contains too much dissolved air gases that may slowly release the excess gases due to gas solubility change in response to slight temperature changes, forming numerous tiny gas bubbles on the surfaces of the water chambers including the Al-Tf-Al membrane surface. These tiny gas bubbles can sometime be so problematic that they could negatively affect the formation and detection of localized protons on the Al-Tf-Al membrane surface because the gas bubbles apparently reside at the water-membrane interface and form an air-gap barrier between the membrane and the liquid water phase. To eliminate this problem for improving the reproducibility of the experiments, a special effort was made in the laboratory water source: the Millipore water was degassed by boiling the water through autoclave and then cooled down to room temperature before the experimental use.
As part of the effort in tracking the integrity of the center chamber assembly’s fitting with the middle O-ring fitting channel of the inter-chamber wall and the sealing at the two ends of the center chamber with the Al-Tf-Al membranes, the conductivity of the water in each of the anode chamber and the cathode chamber was measured after each experiment (Tables S11, S12, S15 and S16). The conductivity measurements for both the anode and the cathode water chambers were in the range from (1.004 ± 0.057) to (2.961 ± 1.130) µS which are acceptable values for pure water conductivity after equilibration with atmospheric carbon dioxide. This means that the possible salt leakage from the Teflon center chamber was negligible in our experiments and that the sealant was tight enough to keep the salt solutions trapped within the Teflon center chamber. Electrolytic current was also monitored to ensure no significant salt leakage from the Teflon center chamber. The observed current in the given set up was in the range from 50 to 70 µA. The experiments were redone again if any leakage was observed i.e.: if the current measurement possessed a magnitude of over 100 µA.
The reason for using bicarbonate salt solutions in our cation exchange experiments was because it was noticed that the proton-sensing film material (aluminum membrane) was sensitive not only to protons but also to a number of other chemical species including Cl–, NO3–, SO4–2, and CH3COO–. For example, it was reported that chloride ions have a high penetration power into the passive aluminum oxide film that protects the aluminum from corrosion (41, 42). This was attributed to its small size that is close to the oxygen atoms in the oxide layer and its high mobility that makes it capable of substituting for the oxygen atoms in the alumina network. Eventually, this may lead to a decrease in the film’s resistivity and hence the corrosion of the aluminum atoms that are beneath the protective layer (43). Similarly, it was reported that aggressive anions like chlorides, thiocyanate, hydroxide, sulfide, nitrate, formate and acetate are highly corrosive to aluminum (44).
It is also important to use freshly prepared sodium bicarbonate solution because the pH of bicarbonate solution differs depending on the concentration and temperature as well as its exposure to air. Increasing the concentration of the bicarbonate results in a decrease in the pH of the solution from 8.40 ± 0.00 (10 mM of sodium bicarbonate) to 8.21 ± 0.01 (700 mM of sodium bicarbonate). Moreover, exposure to atmospheric air, or excessive stirring, or being at relatively higher temperature (as in summer season) enhance the loss of CO2 from the bicarbonate solution to the air. When the sodium bicarbonate solution loses CO2, the solution becomes more alkaline and its pH increases (39).
In our experiments, the sodium bicarbonate was trapped inside the Teflon center chamber where there was no contact with the atmospheric air. This was to ensure that the only factor that affects the corrosive activity on the aluminum surface is the electrostatic localized proton attack on P′ site in the center chamber and not the change in the pH of the solution due to the loss of its carbon dioxide content. A control experiment was performed to evaluate the effect of exposure of bicarbonate solution to the atmospheric air by introducing the bicarbonate solution in an open beaker. The bicarbonate solution (10 mM NaHCO3) that was in contact with atmosphere lost some of its carbon dioxide content and accordingly its pH changed from an initial pH of 8.40 ± 0.00 to 8.86 ± 0.00 after 10 hours.
Another control experiment was performed to evaluate the effect of exposure of bicarbonate solution to the atmospheric air by introducing 10 mM of sodium bicarbonate that was freshly prepared (with initial pH 8.40 ± 0.00) in the following manner and left for 10 hours: 1) Inside the Teflon center chamber that was sealed at both ends with Al film along with a small piece of Al film that was suspended inside, and 2) In a small glass beaker where pieces of Al film were placed on the surface and suspended in the bulk of the solution. After 10 hours, the sodium bicarbonate solution that was trapped inside the Teflon center chamber had a pH of 8.42 ± 0.01 which is close to the initial pH, while the pH of sodium bicarbonate solution in the open beaker rose to a pH of 8.78 ± 0.00. It was also observed that the aluminum pieces inside the Teflon center chamber experienced no changes, while all the aluminum pieces with sodium bicarbonate solution exposed to the air in the open beaker had observable corrosion on their surfaces (Figure S2). This observation indicates that sodium bicarbonate solution which is enclosed in a chamber or a bottle preserves its pH and accordingly no Al corrosion will be observed while sodium bicarbonate solution which is exposed to air for hours loses its carbon dioxide content and accordingly its pH rises to a higher value that could enable Al corrosion.
It was also observed that the temperature has an effect on the pH of the bicarbonate solution (45-47). Sodium bicarbonate solution (10 mM) that was kept in a beaker at 16 ºC had a slightly different pH (8.66 ± 0.00) from the freshly prepared solution (pH 8.40 ± 0.00) and resulted in slightly less corrosive effect on the aluminum pieces than that of the pH 8.86 ± 0.00 sodium bicarbonate solution that was kept in a beaker at room temperature 26 ºC (Figure S2).
Under our experimental conditions we have observed a slight change in the pH of the bicarbonate salt solutions due to different concentrations (Table 3 and Table 4). Using a pH glass electrode would only detect the pH of the bulk medium without detecting the pH changes that may have occurred at the membrane surface. Based on our experimental results in absence of salt, the surface pH of the Al membrane with electrostatically localized protons was well below pH 4 as it was observed by the corrosion activity on P′ side (Table 2, row 1-6). However, salt addition into the center chamber induced an increase in the surface pH at the P′ site which is directly proportional to the concentrations of the cations added through cation exchange with the localized protons as discussed above. Consequently, altering the electrolyte composition and/or concentration of the bulk medium would cause significant changes in the local pH at the membrane surface with only minimal altering in the bulk-phase pH of the bulk solution. These observations may have implications also in understanding the salinity tolerance in biological systems in relation to localized proton coupling bioenergetics.
Experimental Implications on Other Proton Localization Hypotheses and Anomalies to Mitchell’s Chemiosmotic Theory
Notably, the proton-cation exchange equilibrium constants as experimentally determined from our study reported above can now help explain some of the observations previously made by Dilley’s group in their thylakoids experiments (48, 49), in which localized proton coupling was demonstrated in their “low salt” (<10 mM K+) treated thylakoids while delocalized proton behavior was observed in “high salt” (100 mM K+) treated samples. As shown in Table 2, the midpoint of K+ cation exchange with the localized proton layer is at the potassium (K+) bicarbonate concentration of 50 mM. With the use of 10 mM potassium (K+) bicarbonate solution, the localized protons concentration [HL+ ] was essentially not affected as detected by a proton-sensing film placed at the cathode (P′) site in contact with potassium bicarbonate solution. This result is in an excellent agreement with the localized proton coupling characteristics observed in the “low salt” (<10 mM K+) treated thylakoids of the Chiang-Dilley experiment (48). The data in Table 2 also showed that when the concentration of potassium (K+) bicarbonate solution was increased to 100 mM, a significant amount of the localized protons [HL+] was delocalized through the cation (K+ )-proton (H+) exchange into the bulk liquid phase, which can also excellently explain the delocalized proton coupling observed in the “high salt” (100 mM K+) treated thylakoids of the Chiang-Dilley experiment (48) as well. Therefore, the experimental results from our “cathode water membrane (Al-Tf-Al) water membrane (Al-Tf-Al) water anode” biomimetic system are remarkably consistent with those made with the biological samples in the Chiang-Dilley thylakoids experiment (48).
Our experimental results, including the localized protons demonstrated at the liquid-membrane interface, are consistent with the ∆pH surface component of proton motive force in ATP synthesis of mitochondria as previously measured by inserting pH-sensitive fluorescein-phosphatidyl-ethanolamine into mitoplast surface (50) and are consistent also with the recently reported lateral pH gradient between OXPHOS complex IV and F0F1 ATP-synthase in folded mitochondrial membranes (51). According to the Lee proton electrostatic theoretical model (22), the localized excess proton layer at the water-membrane interface is likely to be a special monolayer with a thickness probably of about 1 nm, which could therefore be detected by membrane surface probes such as a pH-sensitive fluorescent molecular probe (50) when placed at the water-membrane interface. A recently reported biomimetic experimental study (52) by the Pohl group using a pH-sensor fluorescein that was covalently linked to N-(fluorescein-5-thiocarbamoyl)1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine and a caged-proton compound now appears also pointing to a similar localized proton phenomenon; their experiments “reveal an entropic trap that ensures channeling of highly mobile protons along the membrane interface in the absence of potent acceptors”. This also indicates that the fundamental physical chemistry of proton-cation exchange with localized protons including the proton-cation exchange equilibrium constants as determined from our “cathode water membrane (Al-Tf-Al) water membrane (Al-Tf-Al) water anode” biomimetic system is probably quite universal, which may be applicable to the study of biological systems (49-51, 53).
In their effort to explain the “low salt” (<10 mM K+) vs. “high salt” (100 mM K+) treated thylakoids experimental observation (48) (which we now know is clearly valid since we were also able to demonstrate essentially the same effect of K+ cations on localized protons in our biomimetic experiments reported above), Dilley et al. (48, 49, 54) conjectured that a hypothetical proteins-occluded space along the membrane surface could provide a localized proton pathway to the ATP synthase, but no evidence for such a proteins-occluded space system has ever been found. Similarly, other conjectured explanations for protons being localized at membrane surfaces such as the “localized proton microcircuits” by Branden et al. (55) and by Liu et al. (56) have not gained acceptance. None of those proton localization models could be supported by our experimental results that demonstrated proton electrostatic location at the liquid-membrane interface forming the anions-membrane-proton capacitor-like structure. Based on our experimental results, it is not necessary to use any of the “hypothetical proteins-occluded space along the membrane surface” (49), the “interaction between the Cytochrome caa3 and F1F0-ATP Synthase” (56) or the phospholipids “proton-collecting antenna” (55) to explain the localized proton coupling in biological systems. As shown with the data of Table 2, the electrostatic localization of excess protons can occur with the use of a proton-impermeable membrane made of non-protein and non-lipid material including Teflon and Al films without any protein and/or any phospholipids (33).
Cherepanov and Mulkidjanian et al. (2006) proposed an interfacial barrier model (57), which used the dielectric permittivity of interfacial water (58). According to their model, there would be a potential barrier of about 0.12 eV for protons in the water phase some 0.5–1 nm away from the membrane surface. That potential barrier was thought to retard the proton exchange between the membrane surface and the bulk aqueous phase thus causing an elevation of the proton concentration at the interface. However, if that is true, the excess protons generated by the electrolytic anode electrode in the bulk phase as demonstrated in our experiment would not be able to enter into the liquid-membrane interface, since the putative “potential barrier” (if exists) between the bulk phase and liquid-membrane interface would have prevented the entry of any excess protons from the bulk liquid phase into the liquid-membrane interface. This prediction from the interfacial barrier model is in contrast to our experimentally observed excess proton electrostatic localization at the liquid-membrane interface as shown in Table 2. Therefore, our study finds no support for the putative interfacial barrier model (57) that anyhow is not really required to explain the localized proton coupling bioenergetics (22) either.
Conclusion
Our experimental results showed again that the Mitchellian proton delocalization view is not true. Both the proton-sensing film detection and bulk liquid pH measurement have now demonstrated, for the first time, that protons can be localized at a water-membrane interface through electrostatic induction at the P′ site in a “cathode water membrane (Al-Tf-Al) water membrane (Al-Tf-Al) water anode” system where the third water body is placed in between an anode water chamber and a cathode water chamber interacting in series. The electrostatically localized excess protons are distinctly different from the fixed-charge-attracted electric double layer phenomenon. The Lee proton electrostatic localization model (22) predicts that the localized excess protons are likely to be in a monolayer at the water-membrane interface that may be exchanged with the non-proton cations in the liquid.
Our experimental results demonstrated that there is an inverse proportionality between the concentration of the salt solution and the proton-sensing corrosion activity of the proton sensing Al film placed at P′ site. By increasing the salt concentration inside the small Teflon center chamber, the proton-sensing corrosion activity of the aluminum membrane placed at P′ site would decrease until showing no proton-associated corrosion activity when the salt concentrations are above 200 mM for both sodium and potassium salt solutions. This was attributed to the delocalization of the localized protons at the membrane-water interface through cation exchange by the added cations of the salt solution.
According to the Lee proton electrostatic localization hypothesis (21, 22), the equilibrium constant for protons to electrostatically occupy the cation sites at the water-membrane interface (in any possible competition with any other cations) is likely to be extremely larger than one. Conversely, the equilibrium constant KPi for non-proton cations such as Na+ to delocalize the localized protons from the membrane-water interface is expected to be extremely smaller than one. Through our experiments reported above, we have now experimentally determined for the first time that the equilibrium constant for non-proton monovalent cations to exchange with the electrostatically localized protons is indeed much less than one (likely on the order of 10-8). The equilibrium constant KPNa+ for sodium (Na+) cations to exchange with the electrostatically localized protons was determined to be (5.07 ± 0.46) x 10-8. Similarly, the equilibrium constant KPi for potassium (K+) cations to exchange with the electrostatically localized protons was determined to be (6.93 ± 0.91) x 10-8. These results mean that the electrostatically localized protons at the water-membrane interface are so stable that it requires of the order of ten million more sodium (or potassium) cations than protons in the bulk liquid phase to even partially delocalize the localized protons at the water-membrane interface. This provides a logical experimental support of the proton electrostatic localization hypothesis (21, 22). It may also have fundamental implications in understanding the salinity tolerance in biological systems in relation to localized proton coupling bioenergetics.
Acknowledgement
This work was supported in part with the Lee laboratory start-up research funds that were provided by the Department of Chemistry and Biochemistry, the College of Sciences, the Office of Research at Old Dominion University, and by the Old Dominion University Research Foundation.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions
J.W.L. designed research and experiments, analyzed data and wrote the article; H.A.S. performed experiments, analyzed data and wrote the article under the guidance of J.W.L.
Supporting Information
Detailed images, pH and conductivity measurements can be found in supporting information. This material is available free of charge at the journal website.
References
1. Mitchell, P. (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144-148
2. Mitchell, P. (1976) Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol 62, 327-367
3. Mitchell, P. (1978) David Keilin’s respiratory chain concept and its chemiosmotic consequences. Nobel prize lecture 1, 295-330
4. Garrett, R., and Grisham, C. (2013) Biochemistry, Brooks/Cole
5. Nelson, D. L., Lehninger, A. L., and Cox, M. M. (2013) Lehninger principles of biochemistry, Macmillan
6. Nicholls, D. G., and Ferguson, S. (1992) Bioenergetics 2, New York: Academic Press
7. Vinkler, C., Avron, M., and Boyer, P. D. (1978) Initial formation of ATP in photophosphorylation does not require a proton gradient. FEBS letters 96, 129-134
8. Ferguson, S. J. (1985) Fully delocalised chemiosmotic or localised proton flow pathways in energy coupling?: A scrutiny of experimental evidence. Biochimica et Biophysica Acta (BBA)-Reviews on Bioenergetics 811, 47-95
9. Dilley, R. A., Theg, S. M., and Beard, W. A. (1987) Membrane-proton interactions in chloroplast bioenergetics: Localized proton domains. Annual review of plant physiology 38, 347-389
10. Dilley, R. A. (2004) On why thylakoids energize ATP formation using either delocalized or localized proton gradients–a Ca2+ mediated role in thylakoid stress responses. Photosynth Res 80, 245-263
11. Williams, R. (2011) Chemical advances in evolution by and changes in use of space during time. J Theor Biol 268, 146-159
12. Krulwich, T. A. (1986) Bioenergetics of alkalophilic bacteria. The Journal of Membrane Biology 89, 113-125
13. Krulwich, T. A., and Guffanti, A. A. (1989) Alkalophilic bacteria. Annual Reviews in Microbiology 43, 435-463
14. Olsson, K., Keis, S., Morgan, H. W., Dimroth, P., and Cook, G. M. (2003) Bioenergetic properties of the thermoalkaliphilic Bacillus sp. strain TA2. A1. J Bacteriol 185, 461-465
15. Sturr, M. G., Guffanti, A. A., and Krulwich, T. A. (1994) Growth and bioenergetics of alkaliphilic Bacillus firmus OF4 in continuous culture at high pH. J Bacteriol 176, 3111-3116
16. Krulwich, T. A. (1995) Alkaliphiles:‘basic’molecular problems of pH tolerance and bioenergetics. Mol Microbiol 15, 403-410
17. Padan, E., Bibi, E., Ito, M., and Krulwich, T. A. (2005) Alkaline pH homeostasis in bacteria: new insights. Biochimica et biophysica acta (BBA)-biomembranes 1717, 67-88
18. Guffanti, A., and Krulwich, T. (1984) Bioenergetic problems of alkalophilic bacteria. Biochem Soc T 12, 411
19. Krulwich, T. A., Gilmour, R., Hicks, D. B., Guffanti, A. A., and Ito, M. (1998) Energetics of alkaliphilic Bacillus species: physiology and molecules. Advances in microbial physiology 40, 401-438
20. Krulwich, T. A., Liu, J., Morino, M., Fujisawa, M., Ito, M., and Hicks, D. B. (2011) Adaptive mechanisms of extreme alkaliphiles. Extremophiles handbook, 119-139
21. Lee, J. (2012) Proton-electrostatics hypothesis for localized proton coupling bioenergetics. Bioenergetics 1, 104
22. Lee, J. (2015) Proton-electrostatic localization: explaining the bioenergetic conundrum in alkalophilic bacteria. Bioenergetics 4, 121
23. Marx, D., Tuckerman, M. E., Hutter, J., and Parrinello, M. (1999) The nature of the hydrated excess proton in water. Nature 397, 601-604
24. Pomès, R., and Roux, B. (2002) Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophysical Journal 82, 2304-2316
25. Marx, D. (2006) Proton transfer 200 years after von Grotthuss: Insights from ab initio simulations. Chemphyschem 7, 1848-1870
26. McLaughlin, S. (1977) Electrostatic potentials at membrane-solution interfaces. Current topics in membranes and transport 9, 71-144
27. Kell, D. B. (1979) On the functional proton current pathway of electron transport phosphorylation: an electrodic view. Biochimica et Biophysica Acta (BBA)-Reviews on Bioenergetics 549, 55-99
28. Hiemenz, P. C., and Rajagopalan, R. (1997) Principles of Colloid and Surface Chemistry, revised and expanded Vol. 14, CRC press
29. Israelachvili, J. N. (2011) Intermolecular and surface forces: revised third edition, Academic press
30. Grahame, D. C. (1947) The electrical double layer and the theory of electrocapillarity. Chemical Reviews 41, 441-501
31. McLaughlin, S. (1989) The Electrostatic Properties of Membranes. Annual Review of Biophysics and Biophysical Chemistry 18, 113-136
32. Lee, J. W. (2013) Membrane surface charges attracted protons are not relevant to proton motive force. Bioenergetics 2, e114
33. Saeed, H. A., and Lee, J. W. (2015) Experimental Demonstration of Localized Excess Protons at a Water-Membrane Interface Bioenergetics 4, 127
34. Pourbaix, M. (1974) Atlas of electrochemical equilibria in aqueous solutions.
35. Pourbaix, M. (1974) Applications of electrochemistry in corrosion science and in practice. Corrosion Science 14, 25-82
36. Eigen, M., and De Maeyer, L. (1958) Self-dissociation and protonic charge transport in water and ice. Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences, 505-533
37. Vargel, C. (2004) Corrosion of aluminium, Elsevier
38. Bell, W. A. (1962) Effect of calcium carbonate on corrosion of aluminium in waters containing chloride and copper. Journal of Applied Chemistry 12, 53-55
39. D.P. Verdonik, R. L. D., F.W. Williams. (1999) U.S. Navy Halon 1211 Replacement Program: Assessment of Aircraft Collateral Damage from Dry Chemical Fire Extinguishing Agents. Naval Research Laboratory, Washington, DC
40. Scamans, G. M., Birbilis, N., and Buchheit, R. G. (2010) 3.08 – Corrosion of Aluminum and its Alloys. In Shreir’s Corrosion, pp. 1974-2010 (Editor-in-Chief: Tony, J. A. R., ed), Elsevier, Oxford
41. Heine, M., Keir, D., and Pryor, M. (1965) The specific effects of chloride and sulfate ions on oxide covered aluminum. Journal of The Electrochemical Society 112, 24-32
42. Britton, S. C., and Evans, U. R. (1930) CCXXXIII.—The passivity of metals. Part VI. A comparison between the penetrating powers of anions. Journal of the Chemical Society (Resumed), 1773-1784
43. Dacres, C. M. (1977) An Investigation of the Influence of Various Environmental Factors Upon the Aqueous Corrosion of Aluminum Alloys, American University
44. Afzal, S., Shaikh, M. A., Mustafa, C., Nabi, M., Ehsan, M., and Khan, A. (2007) Study of Aluminum Corrosion in Chloride and Nitrate Media and its Inhibition by Nitrite. Journal of Nepal Chemical Society 22, 26-33
45. Barron, J., Ashton, C., and Geary, L. (2006) The effects of temperature on pH measurement. In 57th Annual Meeting of the International Society of Electrochemistry, Edinburgh, Technical Services Department, Reagecon Diagnostics Ltd, Ireland
46. Galster, H. (1991) pH measurement: fundamentals, methods, applications, instrumentation, VCH
47. Galster, H. (2000) pH Measurement and Control. In Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA
48. Chiang, G. G., and Dilley, R. A. (1989) Intact Chloroplasts Show Ca-2+-Gated Switching between Localized and Delocalized Proton Gradient Energy Coupling (Atp Formation). Plant Physiol 90, 1513-1523
49. Dilley, R. A., Theg, S. M., and Beard, W. A. (1987) Membrane-Proton Interactions in Chloroplast Bioenergetics – Localized Proton Domains. Annu Rev Plant Phys 38, 347-389
50. Xiong, J. W., Zhu, L. P., Jiao, X. M., and Liu, S. S. (2010) Evidence for Delta pH surface component (Delta pH(S)) of proton motive force in ATP synthesis of mitochondria. Bba-Gen Subjects 1800, 213-222
51. Rieger, B., Junge, W., and Busch, K. B. (2014) Lateral pH gradient between OXPHOS complex IV and F0F1 ATP-synthase in folded mitochondrial membranes. Nat Commun 5
52. Weichselbaum, E., Osterbauer, M., Knyazev, D. G., Batishchev, O. V., Akimov, S. A., Nguyen, T. H., Zhang, C., Knor, G., Agmon, N., Carloni, P., and Pohl, P. (2017) Origin of proton affinity to membrane/water interfaces. Sci Rep-Uk 7
53. Preiss, L., Klyszejko, A. L., Hicks, D. B., Liu, J., Fackelmayer, O. J., Yildiz, O., Krulwich, T. A., and Meier, T. (2013) The c-ring stoichiometry of ATP synthase is adapted to cell physiological requirements of alkaliphilic Bacillus pseudofirmus OF4. P Natl Acad Sci USA 110, 7874-7879
54. Dilley, R. A. (2004) On why thylakoids energize ATP formation using either delocalized or localized proton gradients – a Ca2+ mediated role in thylakoid stress responses. Photosynth Res 80, 245-263
55. Branden, M., Sanden, T., Brzezinski, P., and Widengren, J. (2006) Localized proton microcircuits at the biological membrane-water interface. P Natl Acad Sci USA 103, 19766-19770
56. Liu, X., Gong, X., Hicks, D. B., Krulwich, T. A., Yu, L., and Yu, C.-A. (2007) Interaction between the Cytochrome caa(3) and F(1)F(0)-ATP Synthase of Alkaliphilic Bacillus pseudofirmus OF4 is Demonstrated by Saturation Transfer Electron Paramagnetic Resonance and Differential Scanning Calorimetry Assays. Biochemistry-Us 46, 306-313
57. Mulkidjanian, A. Y., Heberle, J., and Cherepanov, D. A. (2006) Protons @ interfaces: Implications for biological energy conversion. Bba-Bioenergetics 1757, 913-930
58. Cherepanov, D. A., Feniouk, B. A., Junge, W., and Mulkidjanian, A. Y. (2003) Low dielectric permittivity of water at the membrane interface: Effect on the energy coupling mechanism in biological membranes. Biophysical Journal 85, 1307-1316
Figures and Tables
Table 1. Three replicates of final pH measurements for experiments with arrangement “cathode water-Al-Tf-Al-DI water- Al-Tf-Al- water anode” after 10 hours electrolysis (see Table S1-S2).
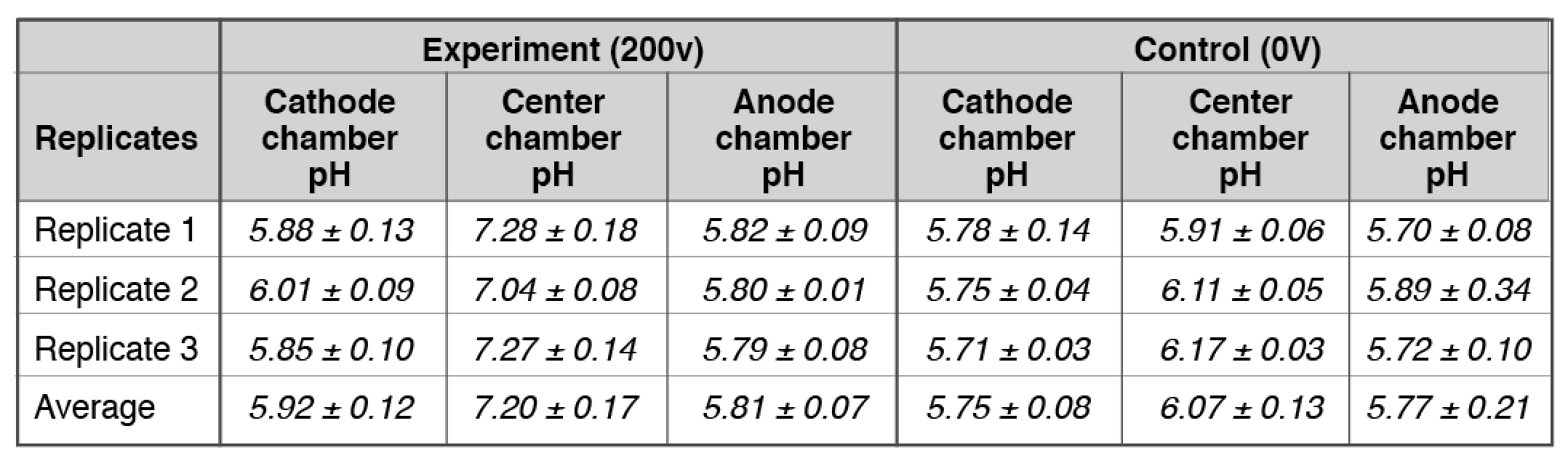
Table 2. Observation of proton-sensing films after 10 hours of electrolysis (200 V) for the “cathode water Al-Tf-Al- bicarbonate solution – Al-Tf-Al water anode” experiment. Images show proton-sensing films that were placed at P′ sites for both sodium and potassium bicarbonate solutions. (See Table S3–S8 for more detailed data)

Table 3. pH measurements for a series of concentrations of freshly prepared sodium salt solution inside the Teflon center chamber after 10 hours open-circuit electrolysis at 200V.
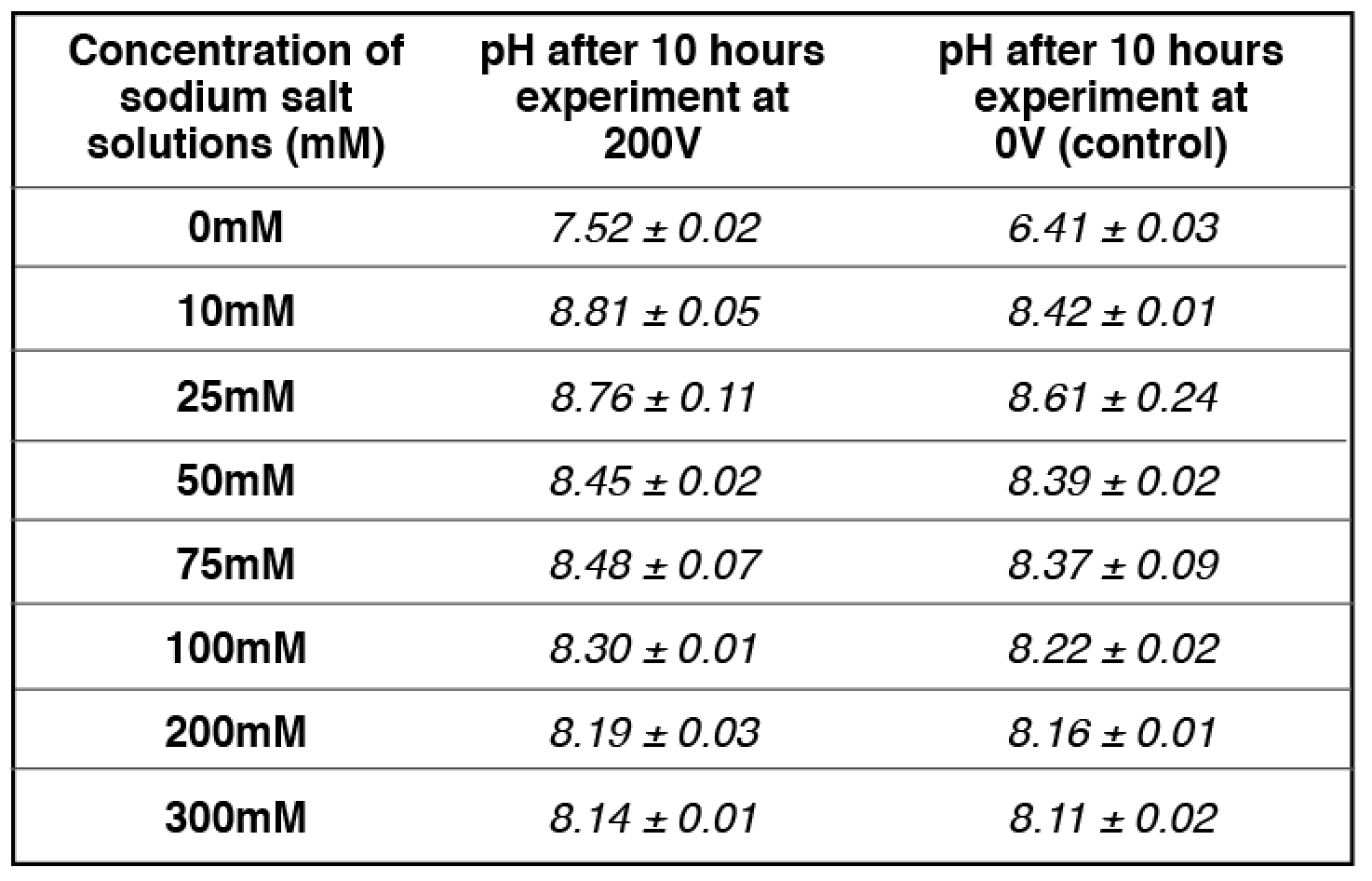
apH measurement for 75 mM Sodium bicarbonate (midpoint with 50-50% sodium/proton exchange) is average of 4 replications while the rest of pH measurements are averages of 2 replications. See Tables S9-S12, S17 for more detailed information.
Table 4. pH measurements for a series of concentrations of freshly prepared potassium salt solution inside the Teflon center chamber after 10 hours open-circuit electrolysis at 200V.
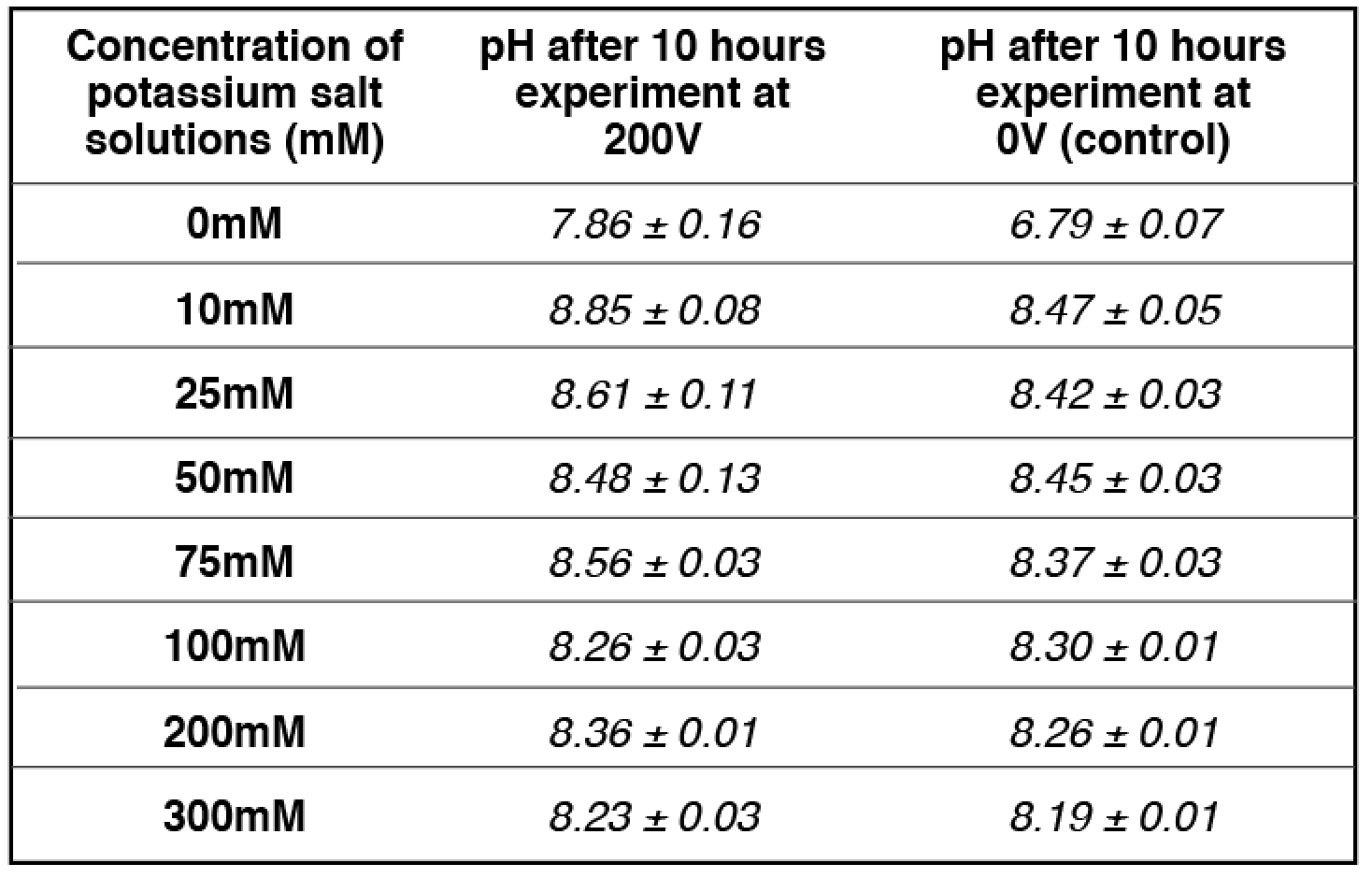
apH measurement for 50 mM potassium bicarbonate (midpoint with 50-50% potassium/proton exchange) is average of 4 replications while the rest of pH measurements are averages of 2 replications. See Tables (S13- S16, S18) for more detailed information.
Figure 1. Schematic diagram showing experimental demonstration of a localized excess protons layer at the water-membrane interface in an “anode water-membrane-water cathode” system. Top (a): showing the excess proton monolayer is extended from a secondary proton layer of the “electric double layer” that covers the anode surface when electrolysis voltage is applied; Bottom (b): showing the likely distribution of excess protons and excess hydroxyl anions in the two water chambers separated by a membrane when electrolysis voltage is turned off.
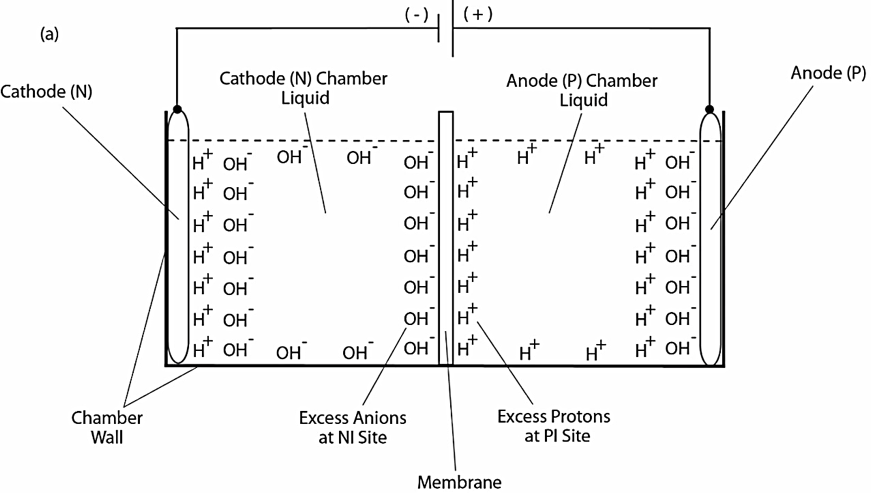
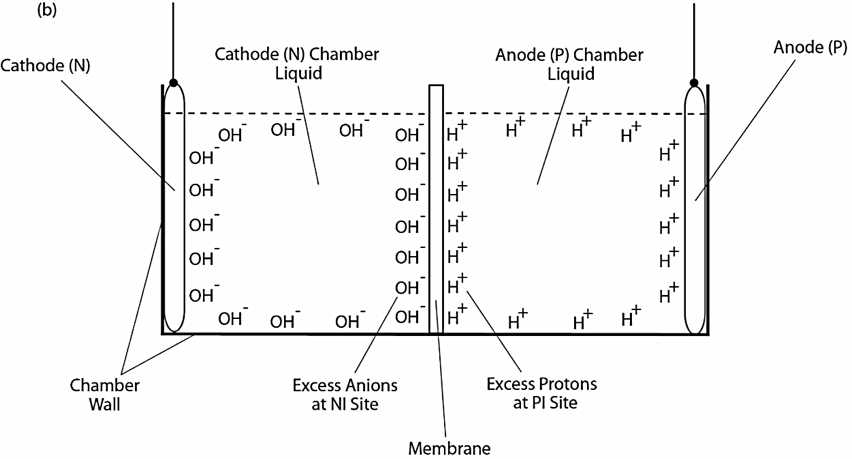
Figure 2. Schematic diagram of the system testing the effect of sodium cations on localized protons at the P′ side in the Teflon center chamber. The inset shows the exchange of the added sodium (Na+) cations with the electrostatically localized protons at the P′ side. A small piece of proton-sensing Al film was placed into the bulk liquid phase (the CB site) of the center chamber.
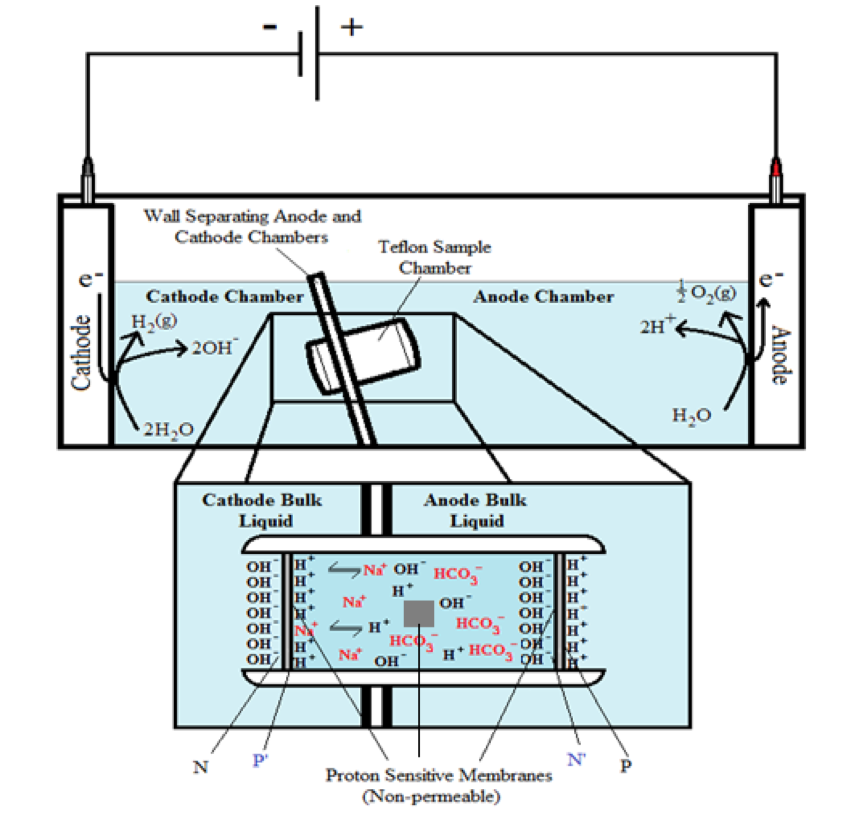
Figure 3. Schematic diagram showing the distribution of protons and hydroxide ions in the cathode, center and the anode water chambers under the influence of applying 200V when the electrodes are polarized. The inset shows the electrostatic distributions of protons and hydroxide ions on P′ and N′ sites respectively in a “water-membrane-water-membrane-water” system.
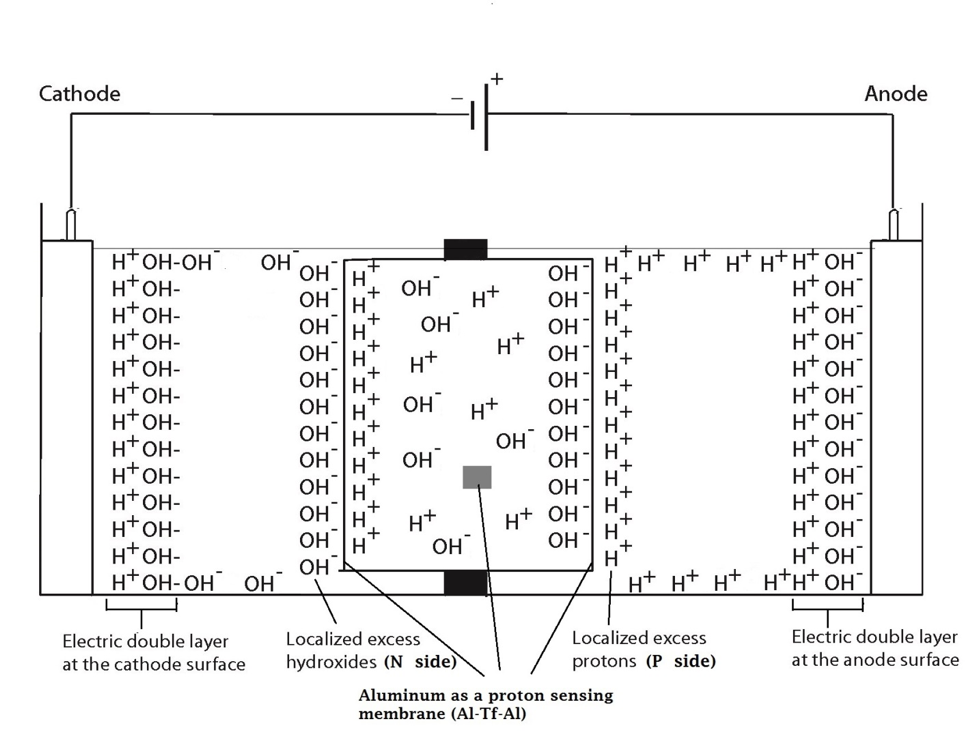
Figure 4. Observation of proton-sensing films after 10 hours of electrolysis (200 V) for the cathode water Al-Tf-Al-DI water- Al-Tf-Al water anode experiment. Images show proton-sensing films that were placed at N, P, N′, P′, NB, PB and CB sites.
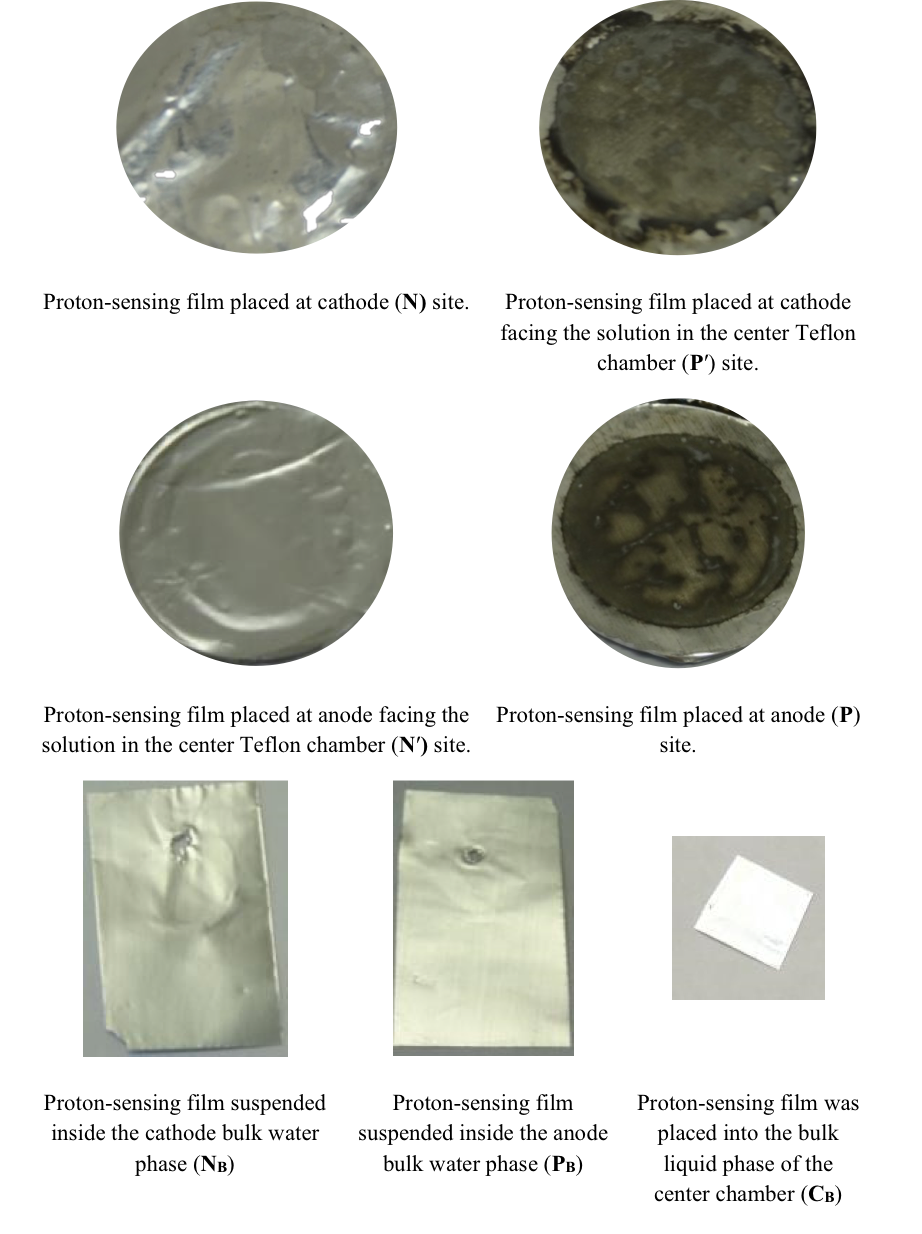
Figure 5. Schematic diagram after introducing salt into Teflon center chamber showing the distribution of different ions in the cathode, center and the anode water chambers under the influence of applying 200V when the electrodes are polarized. The inset shows the exchange of the added sodium (Na+) cations with the electrostatically localized protons at the P′ side in a “water-membrane-sodium bicarbonate-membrane-water” system.